ゼオライトは酸触媒としても触媒担体としても広く利用される材料であり,その分子サイズのナノ空間は,高い比表面積を稼ぐだけでなく,特異な触媒能の根源でもある。ゼオライト学会のウェブサイトでも紹介されているように,ゼオライト細孔(ナノ反応空間)と分子の幾何学的関係によって反応の速度や選択性が影響を受ける「形状選択性」は,他の触媒系には見られないゼオライトに特異な機能である。パラフィンにおけるノルマル体/イソ体の選択性や,キシレンにおけるパラ位選択性がよく知られた代表例である。さらに,ナノ反応空間の形状だけでなく親疎水性等を利用した新たな反応系の開発が進められており,生体触媒の模倣やそれを超えた特異な機能の発現が期待されている。特異な触媒活性の裏には,ゼオライトならではのナノ反応空間に由来する機能が存在し,このナノ反応空間の中で進行する反応を理解することは,長くゼオライト科学における挑戦の一つである。
筆者らは,ナノ反応空間の存在によって進行する反応の一つであるメタノール転換反応に着目してきた。本稿では特に「MTO型反応」と呼ばせていただく。ここで述べる「MTO型反応」とは,ゼオライトおよび類縁物質を酸触媒として用いた,カーボンプールと呼ばれる炭化水素中間体を経由するメタノール転換および類似反応系であり,必ずしも原料および生成物を限定する意図はない。この反応では,ナノ反応空間にカーボンプール中間体を保持する(閉じ込める)ことが鍵であり,反応空間による形状選択性を受けたカーボンプール中間体が触媒性能に強く影響を与えると考えられている。一方で,多種多様な中間体が形成され,複数の反応が並列で進行するため,主たる反応過程の同定とその追跡は困難である。本稿では筆者らの観点に基づいて既往の研究を紹介すると共に,近年取り組んできた定常状態同位体過渡応答法について紹介する。まずは前提となるMTO型反応について,これまでの研究によって明らかになった反応機構等を紹介する。次に,同位体過渡応答法について紹介し,我々の取り組みにおける工夫について示す。最後に,実際に得られた実験結果について解釈する。
2.1 ゼオライト酸触媒を用いたメタノール転換
ゼオライト酸触媒を用いたメタノール転換による炭化水素類合成の歴史は長く,時代の変遷と共に,求められる生成物が変わることで反応系もその名称を変えてきた。最も広い呼称としてはMethanol-to-Hydrocarbons(MTH)と呼ばれている。起こりは1970年台,EXXONによってZSM-5ゼオライト触媒を用いたメタノール転換による高オクタン価ガソリン級成分の合成(Methanol-to-Gasoline (MTG))が検討された。これを受けて広く8–12員環細孔のゼオライトが様々に検討され,ゼオライトの結晶構造(および粒子形状,酸量,酸性質,等)に対して触媒性能(生成物分布,触媒寿命,失活挙動)を評価する研究が行われた1)。1990年台から2000年台初頭にかけ,シェール革命を主とする資源転換とそれに伴うオレフィン類合成プロセスの再検討がなされるなかで,同プロセスを用いた低級オレフィン類の選択的合成が盛んになり,これはMethanol-to-Olefins(MTO)と呼ばれている。特に,プロピレン生成を目的とした場合には,Methanol-to-Propylene(MTP)とも呼ばれる。高いC2–C3オレフィン選択性を示すことから,触媒はSAPO-34に代表される小細孔ケージ型ゼオライト類へと変遷し,現在も盛んに研究が行われている2)。現在,新たな時代の要求としてCO2の利用が求められ,CO2水素化によるメタノール合成と組み合わせる等の複合触媒の開発が検討されている。
2.2 反応機構
目的生成物(および反応の呼称)が変わり,そのためのゼオライト触媒特性(骨格構造,骨格組成,酸量,等)が変更される一方で,基本となる反応メカニズムはそれほど大きくは変わらない。MTGプロセス開発の時代から,生成物であるオレフィンが再び反応サイクルに関与する自己触媒反応挙動の理解を目指し,その反応機構に関する議論が続けられてきた。MTO型反応の概要として,①表面活性種形成および最初のC–C結合形成が議論される反応初期,②カーボンプール中間体が議論される定常反応期,③コーク形成などによる活性点被覆・細孔閉塞による失活期,④そして失活後の触媒の再生があり,これらを区別して議論することが多い。特にMTO型反応を特徴付けているのは,定常反応期におけるカーボンプール中間体の形成であり,これはナノ反応空間を有するゼオライト触媒ならではの挙動であると言える1,2)。
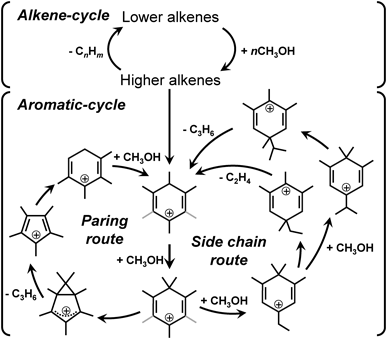
現在広く受け入れられている炭化水素中間体を経由する機構(hydrocarbon pool (HCP) mechanism)はDahlとKolboeによって提唱された。ゼオライト細孔内に形成した(もしくは閉じ込められた)C6–C10程度の比較的大きな反応中間体(カーボンプール中間体)に原料(メタノール等)が取り込まれ反応中間体の一部(または側鎖)が脱離する形で生成物が得られる反応機構のことである(図1)。現在,カーボンプール中間体はアルケン型および芳香族型が広く知られており,これらを媒介してオレフィンが生成する反応系をそれぞれAlkene-cycleおよびAromatic-cycleと呼ぶ。実際の反応ではどちらも進行し,それぞれの寄与度や主たる反応中間体種の違いによって生成物分布が変化すると理解されている。一般には,チャネル型構造のゼオライトではAlkene-cycleを主として比較的長鎖のオレフィン生成物が得られ,ケージ型構造のゼオライトではAromatic-cycleを主としC2–C4オレフィンが得られることが多い。ただし,どちらが優勢に進行するかは,骨格細孔構造だけではなく他のパラメータにも影響されるため,個々の事例については詳細な検討が必要である。近年,中国の研究グループを中心としてSAPO-34の研究が進み,新たにシクロペンタジエンを中間体とするモデルも提案されている3)。
反応機構が非常に独特であるため,時に言葉が一人歩きしがちであるが,ゼオライト上でのメタノール転換は必ずしもMTO型反応(カーボンプールを形成する機構)である必要はないという点は注意が必要である。反応初期のような逐次的なメタノール付加およびクラッキングによるオレフィン生成が支配的なケースや,原料にアルケンが混在する場合には,アルケンへのメタノール逐次付加によるオレフィン生成が主となるケースが報告されている。その一方で,メタノール以外の原料を用いてもMTO型反応は進行する。ジメチルエーテルはメタノールの脱水反応により平衡状態(2CH3OH ⇔ CH3OCH3+H2O)で反応系に存在するため,これを原料として用いても基本的な定常反応挙動は酷似しており,副生する水の影響や,反応初期における挙動の違いなどがより詳細に検討されている4)。また,アルケンを転換する場合においても,カーボンプール種が形成されるMTO型反応となることがある。例えばS. B. HongらはMSE型ゼオライト上でのエチレン転換反応がMTO型反応であることを実験的に示している5,6)。その他,ハロゲン化メチルを原料として用いた場合でも,特定の反応条件下ではMTO型反応が進行することがOlsbyeらによって示唆されている7)。以上を踏まえ,我々はMTO型反応を「ナノ反応空間内に閉じ込められたカーボンプール中間体を媒介して炭素源をオレフィン類へと転換する反応」として広く捉えている。
2.3 MTO型反応とナノ反応空間
カーボンプール中間体およびそれに由来する生成物分布は,反応空間および反応条件によって指向されることがわかっている。同一骨格においても活性点(Al酸点)の骨格内分布が異なれば,活性点周りの反応空間形状に対応した異なる反応中間体が主となる可能性があり,これはゼオライト触媒の特徴である形状選択性が非常に強く発現しているモデルと言える。ケージ型構造においてはAromatic-cycleが優位とされているが,この際にもSide-chainとParingと呼ばれる異なる反応経路が提案されており(図1),これを背景としたケージ選択8–10)・組成調整11,12)・空間デザイン13)が盛んに行われており,一定の成果を挙げている。
一方,広く用いられてきたZSM-5に代表される多次元チャネル型のゼオライトは,細孔部と細孔交点部で異なる広さ・形状の空間を有するため,複数のカーボンプール中間体が存在するモデル(Dual-cycleモデル)が提案され,各反応サイクルの寄与度を制御する試みが行われてきた14–16)。細孔部と細孔交点部における反応をそれぞれ制御しようとする場合,比較的対称性の高いケージ型構造に対し,多次元チャネル型構造では骨格内Alの分布制御および解析をより詳細に行う必要があり,これはゼオライト合成における一つの大きな挑戦である。近年MTO型反応におけるAl分布の影響評価に,Wangら17)および横井ら18)のグループが独自に取り組んだ。骨格内Al分布が異なるZSM-5ゼオライトに対しMTO型反応を行うことで,反応挙動17)や生成物分布18)が変化することを示している。いずれの研究においても,Al分布が異なることによってMTO型反応の挙動・生成物分布に大きな変化が表れたという実験結果を元に,より詳細な検討が推進されたと想像される。一方で,主たる反応経路やカーボンプール中間体が実際には異なっているが,観測された反応挙動や生成物分布の違いが小さいため,見過ごされてきたケースも多々あろうと思う。一般に,特定の構造物性のみを変化させた一連のゼオライトを合成することは難しく,最終的な触媒性能の変化がどのパラメータに起因したものかを実験的に判別すること(構造–性能相関を実験で直接的に示すこと)は基本的に困難である。反応機構の解明・同定には反応中間体・反応経路の解明が必要であり,これにはin-situ分光分析や過渡応答解析が強力なツールとなりうる。次章から我々が取り組んできた同位体を用いた過渡応答解析について述べる。
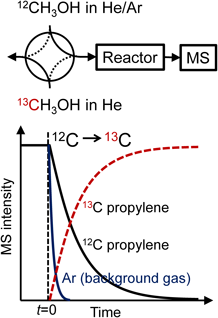
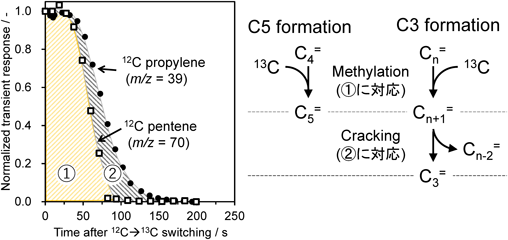
過渡応答解析法は古くから触媒研究に広く使われてきた手法である。反応温度・反応物濃度(もしくは圧力)などのパラメータを切り替えた前後の触媒反応挙動や新たな平衡へ緩和する過程を観察することで,反応に関する情報を引き出すものであり,ジャンプ法とも呼ばれている。基本となる考え方は同じであるが,温度や圧力を変えることで平衡を動かす過渡応答法とは異なり,定常状態を維持したまま同位体標識を導入する過渡応答法を定常状態同位体過渡応答解析(Steady-State Isotopic Transient Kinetic Analysis: SSITKA)と呼ぶ19)。近年においても,FT合成やdeNOxといったホットな研究分野で用いられている20,21)。同位体の応答を観察するため,通常は質量分析器を検出器として用いる19)。図2に,プロピレンを対象としてSSITKAを行う際の装置の概略図と得られるデータを簡単に示す。定常反応を維持したまま,原料として供給する12Cメタノールを13Cメタノールへと高速で切り替える。切り替えのバックグラウンドとして,キャリアガスにAr等の不活性ガスを混合しておく。切り替えと同時に,生成物として得られていた12Cからなるプロピレンが減少し始め,13Cが取り込まれたプロピレンが生成し,この挙動を質量分析器で検出する。
3.1 MTO型反応への同位体過渡応答解析の適用
MTO型反応の解析に同位体標識を用いる試みはBjørgen, Olsbye, Kolboeらの一連の研究によって広く周知されることとなった22)。続くDual-cycleモデルに対するマイルストーンとなる研究では,時間分解能は粗いがSSITKAと類似の解析が行われている23–25)。同位体への切り替えからサンプルを0.5–1分おきに採取し,生成物に含まれる13C量を測定することで,取り込み挙動が似た生成物を分類することに成功している23–25)。この分類の中で,エチレンと芳香族種への同位体取り込み挙動が似ていることから,エチレンはaromatic-cycleから生成しておりalkene-cycleから生成しているより長鎖のアルケン類とは異なる起源であることを見出している。これ以降,同様の解析が幾つかのグループによっても行われてきた。先述したWangらによるAl分布の異なるZSM-5に対するMTO型反応検討においても同様の実験が行われており,パルス的に打ち込まれた13Cメタノール由来の13Cが生成物に取り込まれる挙動を観察している17)。
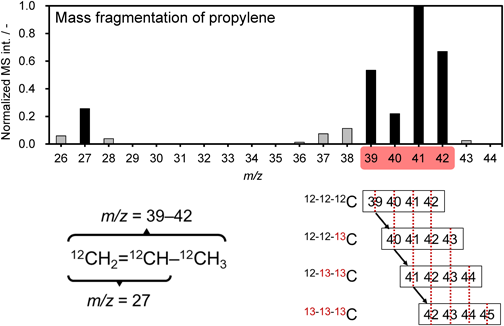
これに対し我々の試みは,より詳細な13C取り込み過程の情報を引き出そうとするものであり,特定の生成物に着目し,13Cが挿入される挙動を追った26)。まず過渡応答実験を行うにあたり,触媒反応系が反応速度解析を行うのに十分な微分反応系であること,反応律速であることを確認した。さらにSSITKAに特有の幾つかの因子(gas-phase holdup効果,同位体効果,クロマトグラフィック効果,等)について補正または無視できるということを確認した。実際の測定に際し,まずはプロピレンをターゲット生成物とした。プロピレンはC3化合物であり,質量電荷比(m/z)が36から43の領域にC–C=C骨格に由来するフラグメントが観察される(図3)。同位体炭素の挿入によってフラグメントの質量電荷比は一つずつ増加し,図3のように変化する。また,質量電荷比27に見られるフラグメントはメチルが脱離したC=C骨格に由来するものである。MTO型反応に対する質量分析法に特有の課題として,複数のオレフィン生成物から同じ質量電荷比を有するフラグメントが多く生成することが挙げられる。プロピレンのC3フラグメントを他の生成物(例えばブテン,ペンテン)から生じた同質量電荷比のフラグメントと区別するため,出口ガスを質量分析器に導入する前に-120℃程度のコールドトラップを設置し,プロピレンより重い成分を回収した。また,測定中にコールドトラップ温度が10℃程度変化するが,これを要因とする過渡応答曲線の変化がないことも確認した。これにより質量電荷比が39から42のフラグメントはプロピレンからのものであることを保証した。
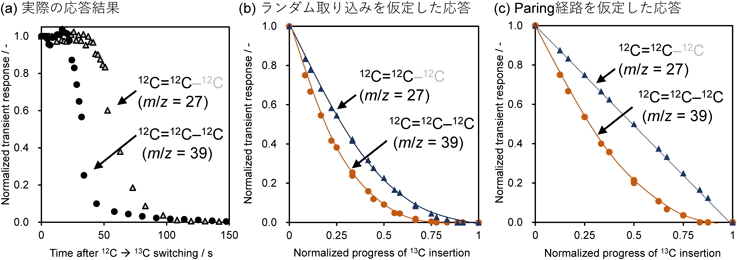
最初に,質量電荷比39および27のフラグメントに着目して分析を行った。図4(a)に実際の測定結果を示す。質量電荷比39のフラグメントの応答は,13Cが生成物であるプロピレン骨格へ最初に取り込まれる挙動を示しており,質量電荷比27の応答はプロピレン中の二重結合部位(C=C骨格)へ13Cが最初に取り込まれる挙動を示している。図4(b)に示すのは,プロピレン骨格に対して13Cが位置選択性を示さずランダムに取り込まれた場合を仮定した際の質量電荷比39(プロピレン骨格)および27(C=C骨格)の応答挙動となる。実際のC=C骨格への13C取り込みは,ランダム取り込みを仮定した場合に比べて遅れていることが比較からわかる。すなわちプロピレン骨格のメチル位(C-C=C)が最初に13Cに置換されている。特に取り込み初期において,質量電荷比39のフラグメントが減少し始めてから27のフラグメントが減少し始めるまでに25 sの遅延が見られる。これをどのように理解するかはまだ議論が十分ではないが,現時点ではプロピレン生成がAlkene-cycle優位に進んでいると解釈される。表1に,各反応機構を仮定した場合に13Cの取り込み初期における位置選択性をまとめて示す。また,その結果想定される質量電荷比39に対する27の減少率の値を示す。Alkene-cycleでは,ゼオライト酸点でメトキシ種となった13Cが,長鎖アルケンの末端二重結合へのメチル化により取り込まれ,続くヒドリド移行反応とβ開裂によってプロピレンが生成する場合,β開裂が起こった側で二重結合が形成されるので,13Cはメチル位(13C-C=C)にのみ取り込まれる。したがって,二重結合側のフラグメントに13Cが取り込まれるのは,13Cが中間体として滞留するようになった後である。これは観測された質量電荷比27のフラグメントの遅延を説明可能であると考えている。一方で,芳香族を中間体とする場合はParingルートでのプロピレン生成挙動を考える必要がある(※Side-chainルートではプロピレン生成には中間体への2分子のメタノール付加が必要となり,プロピレンへの13C取り込みとC=C骨格への13C取り込み挙動は一致すると考えられる)。Paringルートではプロピレン骨格のいずれかの末端に新しい炭素が付加される。これを仮定した場合,図4(c)のような応答となる。ランダム取り込みよりもC=C骨格への13C取り込みが遅れることがわかるが,減少挙動そのものが遅延するわけではない。これらを考え,今回のケースではプロピレン生成はAlkene-cycleを主として進行していると判断している。
表1. 各反応機構における同位体切り替え後のラベル位置パターン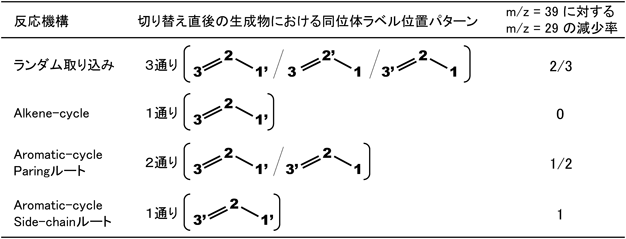 |
続いて,コールドトラップの温度を変えることで,ペンテン(C5)由来のフラグメントとして質量電荷比が70であるものを測定した。これをプロピレン由来フラグメントと比較したものを図5に示す。興味深いことに,プロピレンよりもペンテンへの13C取り込みが相対的に早いということが観測された。これはプロピレンの生成速度が相対的に早いということを示唆している。これまでも述べている通り,プロピレンは「大きな中間体への13C付加→中間体からの生成物脱離」というプロセスを経由する。これに対し,ペンテンはC4中間体への13C付加によっても生成しうる。当然,より大きな中間体への付加→生成物脱離による過程でペンテンが生成する過程を否定することはできないが,β開裂を伴わないC4中間体への13C付加が主生成経路となり,見かけのペンテン生成速度が早いと理解することが自然であろうと思う。この場合,過渡応答の差分がβ開裂に対応することになる。すなわち図5において①で示される区画が付加反応,②の区画がβ開裂にそれぞれ対応する。図より,付加反応に比べてβ開裂の方が早い反応過程であることが示唆されており,この推察はこれまでの検討とも一致している27,28)。今後この仮定を裏付けていく測定が必要であろう。
3.2 Al分布を変えたZSM-5触媒を用いた同位体過渡応答解析
前述したように,カーボンプール中間体はナノ反応空間の形状によって安定化された構造をとると考えられている。したがって,究極的にはカーボンプール中間体は,ゼオライト結晶骨格ではなく,よりミクロな反応空間の形状を反映しているはずである。すなわち,反応が進行する空間の制御によってカーボンプール中間体を制御し,さらには間接的に生成物分布を制御できるということになる。SAPO-34に代表されるケージ型構造を有するゼオライトでは,反応空間は骨格を構成するケージ構造とほぼ同じであるとみなされる場合が多いが,ZSM-5のように細孔部と細孔交差部が存在するようなゼオライトでは,実際の反応空間(すなわちAlが存在する場所)が多岐にわたる。前述の通り,Wangらおよび横井らのグループがAlの分布を制御したZSM-5を調製し,MTO型反応の制御を試みている17,18)。「ゼオライト結晶骨格内のAl分布制御」および「Al分布の正確な測定」はゼオライト科学における重要な課題であり,これらの結果は,ゼオライト構造物性とその性能・機能とを紐付けた一つのマイルストーンと言える。ただし,MTO型反応においては,Al分布と生成物分布が直接関連づいているわけではなく,両者を紐付ける鍵として反応中間体(カーボンプール中間体)の存在がある。我々は,鍵となるこの要素をより直接的に観測することで,他のゼオライト構造物性(結晶サイズ,結晶形状,欠陥量,親疎水性,等)の影響を排除した構造–性能相関を示すことを目的として同位体過渡応答解析を行った26)。
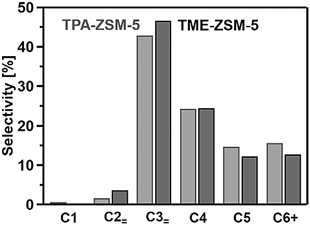
横井らの報告に従って,テトラプロピルアンモニウム(TPA)およびトリメチロールエタン(TME)を用い,異なるAl分布を有する二つのZSM-5ゼオライト(TPA-ZSM-5およびTME-ZSM-5)を調製した。材料評価の詳細は引用文献26を参照していただきたいが,当然,異なる有機物を用いた合成であるので各物性の数値は完全に一致しているとは言えない。特に結晶サイズと結晶形状には大きな違いがあり,触媒作用に一切影響を与えないとは言い切れない26)。制御対象であったAl分布の違いは固体NMR測定によって確認したが,2サンプルの差はそれほど明確ではなかった。得られたNMRスペクトルはブロードなシグナルが多数オーバーラップしており,これを一つ一つの要素まで分解することは困難であった26)。これだけではAl分布の違いを証明するには不十分であると考えられたため,モデル触媒反応によるAl分布の解析を行った26)。ノルマルヘキサンと3-メチルペンタンのクラッキング速度を比較し,炭化水素の大きさと酸点(Al)が分布している空間の大きさとを対応させる手法で,このクラッキング速度比はConstraint Index(CI値)と呼ばれている。我々が調製したZSM-5ゼオライトのCI値はTPA-ZSM-5で1.4, TME-ZSM-5で5.8であった26)。これはTPA-ZSM-5ゼオライトが細孔交差部により多くのAlサイトを有することを示唆している。これとNMRとの結果を合わせてAl分布の異なるZSM-5が合成できたことを確認した18,26)。これらのZSM-5ゼオライトを用いたMTO型反応における生成物分布を図6に示す。二つのZSM-5ゼオライトの生成物分布の違いは,ほぼ同等とも言えるほど小さく,違いは顕著ではなかった。繰り返しになるが,この違いが真にAl分布に起因するか,それとも制御しきれなかった他の構造物性パラメータに起因するものなのか判断するのは一般に非常に困難である。
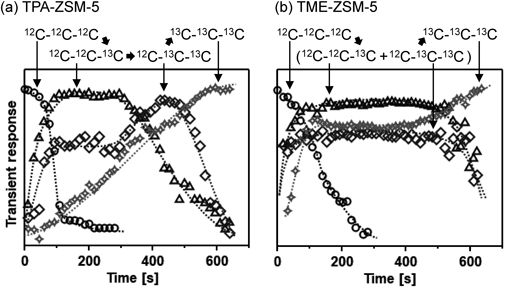
我々は,この違いが真にAl分布に起因していると結論付けるため,SSITKAを行った。図3に示す質量電荷比39から45のフラグメントの応答を観測し,プロピレンへの13Cの取り込み挙動へと解析し直した結果を図7に示す。生成物分布とは異なり,13Cの挿入挙動は二つのZSM-5ゼオライト間で大きな違いを示し,これは異なる反応中間体の存在を強く示唆している。いずれのZSM-5ゼオライトにおいても同位体置換されていないプロピレン(12C–12C–12C)は13Cメタノールへの切り替え後から減少を始め,同位体置換されたプロピレン種(12C–12C–13C, 12C–13C–13C, および13C–13C–13C)が増加し始めた。TPA-ZSM-5では同位体の取り込みが逐次的に進行している(12C–12C–12C ⇒ 12C–12C–13C ⇒ 12C–13C–13C ⇒ 13C–13C–13C)ことが見て取れるが(図7(a)),TME-ZSM-5では12C–12C–13Cおよび12C–13C–13Cのフラグメントがほぼ同じ挙動を示した(図7(b))。これは反応中間体が異なることによる反応機構・反応経路の違いを反映している。
ここまでに示すように,MTO型反応の見かけの生成物分布がほぼ同じゼオライト触媒であっても(図6),Al分布の違いは各生成物の生成挙動の違いとして現れることを実験的に示すことに成功した(図7)。特に,今回測定した同位体炭素の取り込み挙動は,反応中間体もしくは反応メカニズムの違いを直接反映した因子であり,粒子形状や粒子サイズといった構造物性パラメータの影響を受けないと考えらえるため,これまでよりも直接的な構造–性能相関(骨格内Al分布と触媒特性との関係)を示すことに成功したと言える。次の課題は観測された挙動と具体的な反応中間体(もしくは反応経路)とを紐づけることであるが,これには反応中間体のin-situでの同定が必要であり,すなわち究極的な課題の解決となる。