2. 固体NMRによるCO2の分子運動の測定・解析法
細孔内におけるCO2の分子運動は,主に①吸着サイトにおけるローカルな運動,②細孔内の拡散運動,の二つに分けられる。前者は具体的に,吸着サイト上における異方的な回転運動,および吸着サイト間のホッピング運動が存在する。これらの運動にはMOF–CO2間やCO2–CO2間の相互作用,また細孔内の構造が反映され,吸着メカニズムに関する情報を我々に与える。一方,後者の細孔内拡散は吸着サイト間のホッピング運動が長距離にわたって生じたものと解釈することができる。その拡散係数はやはり吸着の強さや細孔径などと強く相関し,ガス透過分離法においては分離能を左右するパラメーターとなる。本章では,こうしたCO2の運動を固体13C NMRによってどのように観測するのかについて説明する。
2.1 固体13C NMR測定の基礎
2.1.1 化学シフト相互作用
NMRは,静磁場中において原子核のスピンを観測する分光法である。核スピンは磁場中において,ゼーマン相互作用によってエネルギー状態の縮退が解けて分裂が生じ,その分裂幅に相当するエネルギーを信号として観測する。実際にはゼーマン相互作用以外にも,核スピンの周囲の環境によって大きさが変わる相互作用がいくつか存在し,これらがNMR信号に構造の情報を反映したピーク位置や線形を与える。固体13C NMRの場合,信号の位置や線形を主に決めるのは,核スピンとその周りの電子が作る磁場との相互作用である化学シフト相互作用である。この化学シフト相互作用は,等方的な成分と異方性を持った成分から構成される。前者は等方化学シフトと呼ばれ,信号の現れる位置を決定する。一方,後者は化学シフト異方性(Chemical Shift Anisotropy; CSA)と呼ばれ,NMRマグネットの静磁場に対する分子の向きによってその大きさが変化する。溶液NMRの場合は溶媒中のランダムな分子運動で後者が平均化され,前者によってピークの位置のみが与えられる。一方,固体の粉末試料では分子・結晶が磁場に対して様々な方向を向くため,その方向に応じた様々な位置に信号が現れる。結果として,スペクトルにはそれらをすべて合算した広幅な信号が観測され,これは粉末パターンと呼ばれる。
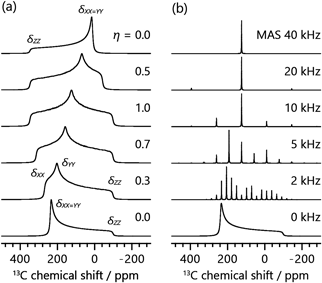
NMRの核スピン相互作用の大きさはテンソル量として表され,相互作用テンソルの主軸座標系においては,3×3の対角行列の対角成分に現れる三つの主値によってその大きさが決定づけられる。化学シフト相互作用では三つの主値(δXX, δYY, δZZ)によって,等方化学シフト値δiso,CSAの大きさδaniso,および異方性の軸対称性を表す非対称性パラメーターηが|δZZ-δiso|≥|δXX-δiso|≥|δYY-δiso|の条件の下,次のように定義される。
これらはスペクトルにおいて,図1aに示すような特徴的な線形を与える。13C核の場合,有機分子では主に結合する原子の種類によって13C核を取り巻く電子雲の形状や密度が変化するために,化学シフトテンソルの主値が変化する。例えば水素三つと結合したメチル基と酸素二つと結合したカルボキシ基では,等方化学シフト値およびCSAパラメーターはともに異なる値を示し,その結果スペクトル上において異なるピーク位置および粉末パターンの形状を与える17)。特にピーク位置については,官能基や化学種によってスペクトル上のどの範囲に信号が現れるかが大方決まっており,構造解析を行う上での重要な情報となっている。
補足として,CSAパラメーターには上述のものとは異なる定義もあるため,文献などを参照する際は注意が必要である。また,NMRでは相互作用の大きさが周波数単位で表されるが,化学シフト相互作用の大きさは静磁場の強さに比例して変化する。そのため,数値を磁場強度に依存せずに取り扱えるよう,化学シフト相互作用の大きさは基準物質のピーク位置で決められた共鳴周波数で割って得られるppm単位の相対的な値によって示される。
2.1.2 マジック角回転法
固体粉末試料の広幅な信号に対し,異方性相互作用を平均化し信号を先鋭化する手法としては,試料管を静磁場に対して54.7°傾けた状態で高速回転するマジック角回転法(Magic Angle Spinning; MAS)が用いられる。MAS法は現代の固体高分解能NMRにおける重要なテクニックの一つであり,専用のMAS NMRプローブおよび回転試料管とともに一般に広く使用されている。図1bに示すように,CSAによる信号の線幅より十分に速いMAS速度を用いれば,異方性を消去し,溶液NMRのような等方化学シフトのみによる細い信号を得ることができる。MAS速度の周波数が線幅よりも小さい場合,等方化学シフトによるピークに加え,MAS周波数に応じた位置にスピニングサイドバンドと呼ばれるサブピークが現れ,結果としてスパイク状の信号を得る。このサイドバンド信号の強度比は,静止試料の粉末パターンの線形と同様,CSAの大きさに応じて変化する。したがって,サイドバンドパターンからCSAパラメーターを抽出することが可能である。
2.2 CO2のローカル運動の測定・解析
2.2.1 化学シフト異方性による線形の解析
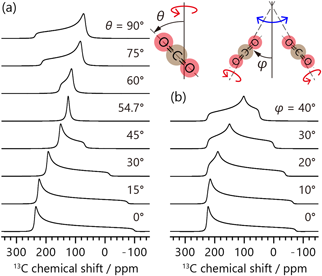
CO2は常温常圧で気体であるため,その13C信号は,CSAがランダムな分子運動で平均化された等方化学シフトのみによるシャープなピークとなる。一方,195 K以下における固体状態ではCSAによる粉末パターンが観測され,そのパラメーターはδaniso=-223 ppm, η=0と報告されている18)。吸着状態においては,吸着サイトとの相互作用や細孔内の立体障害を受けて分子運動が異方的に制限され,13C信号は気体分子でありながらCSAの影響を受けた粉末パターンとなる。この線形を解析することにより,細孔中のCO2の動的な吸着状態を明らかにすることができる。CO2は吸着サイト上において一軸回転運動を行うことが知られ,運動速度を106 Hz以上と仮定したとき,回転軸に対する傾きの角度θに応じて見かけのCSAの大きさδ′anisoが次式のようにスケールされる。
δ0anisoは先述の固体のCO2のCSA値である。室温付近およびより高温においては,運動速度は106 Hz以上としてほぼ問題ないであろう。θの値に応じた粉末パターンの変化の様子を図2aに例示した。式(2)を用いることで,吸着したCO2の粉末パターンの線形より得られるCSAの大きさδ′anisoより,回転運動の傾きの角度θを明らかにできる。また2.1.2で述べたように,遅いMAS速度の下で現れるサイドバンドパターンからもδ′aniso,そしてθを得ることが可能である。δ′anisoを得るための線形解析は,測定データに対するピークフィッティングにより可能である。これには固体NMRスペクトルの解析に対応した専用のソフトウェアが必要であり,筆者はフリーソフトであるssNaKe19)を利用している。もしくは,スペクトルシミュレーションによる解析も可能である。この場合はδ′anisoを得るのではなく,θの値を変えながらスペクトルを計算し,実験結果を再現するθを直接探す作業になる。図2aは実際に固体CO2のCSA値(-223 ppm)を用いて様々なθにおける回転運動下のスペクトルを計算することで得ている。これもやはり専用のソフトウェアが必要であり,フリーソフトであればMATLAB上で動作するEXPRESS20)が使用可能である。
先述の通り,CO2は回転運動に加えて,吸着サイト間をホッピング運動している。このホッピングに伴って結晶構造中における回転運動の軸の向きが変化する場合,回転軸に対する傾きθに加えて,回転軸の角度変化の影響も粉末パターンに現れる。その結果,信号は非対称性パラメーターη≠0の粉末パターンのような線形となる。一例を図2bに示した。このような場合,フィッティングで得られる見かけのCSAパラメーターからθおよび回転軸の角度変化の大きさを求めることはできず,分子運動を考慮したスペクトルシミュレーションによる解析が必要となる。
なお,本稿では化学シフト相互作用のみによって線形が決まる場合を取り扱うが,吸着サイト付近に常磁性を持つ原子がある場合,その電子スピンと13C核との相互作用によっても信号の位置や線形が変化するため,解析が煩雑になる。そのためMOFでは,Zn2+など反磁性金属イオンからなる構造に対してNMR測定・解析が行われる場合が多い。
2.2.2 等方化学シフト値を用いた解析
細孔表面に物理吸着したCO2は,吸着サイトとの静電的相互作用によって13C信号の等方化学シフト値に変化が生じ,そのピーク位置は気体状態と比べてわずかに低周波数(低ppm)側にシフトすることが知られている。気体のピーク位置が125 ppm程度である場合,吸着したCO2の等方化学シフトによるピークの観測範囲は,おおよそ122–125 ppmである。等方化学シフト値は,静止試料の広幅な粉末パターンを解析するより,MASで先鋭化した信号から直接取得する方が高精度に得られやすい。ピークの位置は吸着サイトとの相互作用の強さにある程度依存すると考えられるが,MOFにおいては両者の関係性を系統的に調べ,ピーク位置から相互作用の強さを議論した報告例はなく,等方化学シフトの値そのものから吸着状態を評価することは現状難しい。あるいは,量子化学計算による化学シフトの予測と組み合わせることが有効かもしれない。なお,溶液13C NMRでは一般に標準物質であるテトラメチルシランの信号をスペクトルの0 ppmと定めるが,固体13C NMRではアダマンタンやグリシンなどの2次標準物質を用いて化学シフト値を校正することが多い。どの2次標準物質のどのピークをテトラメチルシランに対して何ppmと設定するかは文献によって少しずつ差があるため,化学シフト値の校正や文献との比較を行う際は注意を要する。また試料中に常磁性原子が存在する場合,先述の常磁性相互作用によって吸着したCO2のピークが気体より高周波数側にシフトしうるため,こちらも注意が必要である21, 22)。
細孔中に異なる複数の吸着サイトが存在し,CO2がそれらの間をNMRの信号観測の時間スケール(13C MAS NMRの場合ms程度)より十分速く行き来している場合,ピークはそれぞれの吸着サイトに対応した位置ではなく,それらを平均化した位置に観測される。この平均的なピーク位置は,各吸着サイトへの吸着量の比で重みづけされる。そのため,吸着等温線を参考にいくつかの圧力で測定を行うことで,ピーク位置の圧力依存性より複数ある吸着サイトがどう埋まっていくのかを解析することができる。
2.2.3 スピン–格子緩和時間を用いた解析
試料を静磁場中に入れた直後やNMR信号の観測直後から磁化,つまりNMR信号が熱平衡状態へと向かう過程はスピン–格子緩和と呼ばれる。磁化Mが熱平衡状態M0に向かって回復する様子は,時定数T1を用いて
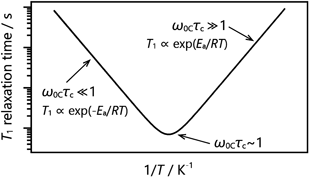
と記述される。実験的には主に飽和回復法や反転回復法によって測定され,これらは現代のNMR分光計には標準的に備わっている測定法である。NMRの緩和は核スピン相互作用によって核が感じる局所磁場が分子運動で揺らぐことによって生じ,局所磁場の揺らぎの速度,すなわち運動の速度に応じて緩和時間は変化する。そのため,試料温度を変化させ分子運動の速度を変えながら緩和時間測定を行い,得られたT1の温度依存性を解析することでその運動性を調べることができる。CO2の13C核では化学シフト相互作用が緩和に寄与し,緩和速度1/T1CSAはBPP式と呼ばれる次の式で表される23)。
CはCSAの大きさと運動モードに依存した定数,ω0Cは13C核の共鳴周波数であり使用するNMR装置の磁場強度による。τcは運動の相関時間,つまり運動速度の逆数であり,大抵アレニウス式τc=τ0 exp(Ea/RT)で記述される。式(4)は温度の逆数に対して図3のような曲線となり,実験で得たT1に対してC,τ0, Eaを変数としてフィッティングを行い解析する。これより,運動の活性化エネルギーEaと運動速度1/τcが求められる。詳細は説明しないが,Cから運動モードを解析することも可能である。なお,T1の値はω0Cτc ~1において図3のT1曲線を特徴づける極小値を取るが,運動の速度や実験条件によっては極小値を観測できないことがある。この場合,式(4)を解析に使うことは難しいが,運動の速度1/τcが共鳴周波数ω0Cより十分遅い領域では,ω0Cτc≫1より式(4)を近似した
逆に運動が共鳴周波数ω0Cより十分速い領域では,ω0Cτc≪1より
によって解析を行うことができる。ただしこれらの式によるフィッティングでは,Cとτ0はそれらの積としてしか得られず,したがって明らかになるのはEaのみである。
MOFのように細孔中に水素原子が多く存在する場合,その1H核とCO2の13C核との間に,大きさが核間距離に依存する双極子相互作用が働く。化学シフト相互作用と同様に双極子相互作用も緩和に影響し,その緩和速度1/T1dipは
と与えられる23)。Dは双極子相互作用の大きさとCO2の運動性に依存し,ω0Hは1H核の共鳴周波数である。またCO2の13C核のT1は,これら二つの相互作用による緩和速度の和(1/T1CSA)+(1/T1dip)で与えられる。なお,T1の極小が観測できない場合は,双極子相互作用の寄与の有無にかかわらず,やはり式(5, 6)の近似式で解析することになる。
2.3 CO2の拡散運動の測定・解析
2.3.1 拡散NMR法
CO2を含む様々なガス分子やイオン,溶液中の分子などの拡散係数が拡散NMR法によって測定されている。測定は,NMRマグネットの静磁場に磁場勾配を生じさせて行われる。スペクトルにおけるNMR信号の位置を主に決めるゼーマン相互作用および化学シフト相互作用の大きさは,磁場強度に比例する。そのため,磁場勾配があると試料位置によって磁場強度が変化し,それに応じて信号の位置に違いが生じる。試料中で分子やイオンが動き回る場合,移動距離に応じて信号の位置が変化するので,これを利用することで拡散係数を解析することができる。非常に強力な測定法だが,磁場勾配をかけられる専用のNMRプローブおよび磁場勾配パルスを用いた特殊な測定シークエンスが必要となる。測定・解析法の詳しい説明はここでは割愛するが,詳細を知りたい場合は例えば「竹腰清乃理著:磁気共鳴–NMR―核スピンの分光学―」の10.6節に測定法についてコンパクトにわかりやすく書かれている。また,筑波大学の早水先生がhttps://diffusion-nmr.jp/にて主にLiイオンの拡散NMR測定に関する詳細な解説を公開されており,こちらも非常に参考になるであろう。ちなみにMOF細孔中におけるCO2の拡散係数は,細孔の構造に依存するものの,一般的には10−8–10−10 m2/s程度と拡散NMR法により測定されている24–29)。
2.3.2 交差分極法
交差分極法(Cross Polarization; CP)は,核スピン間の双極子相互作用を利用してある核から別の核に磁化を移動し観測する方法である。固体の有機物の13C測定においては,周囲に多く存在し測定感度のよい1H核から磁化を移すことで,13C核の信号強度を増幅し,測定効率を大幅に向上できる。そのため,MAS法とともに現代の固体NMRの重要なテクニックの一つとなっている。緩和の項で説明したように,MOF細孔中ではCO2の13C核が周囲の1H核と双極子相互作用を形成する。しかしながら,双極子相互作用の大きさは核間距離の3乗に反比例し,吸着したCO2は拡散によって周囲の1Hと距離が離れて双極子相互作用が弱まってしまう。そのため,基本的にはCP法で信号を検出することはできない。筆者の経験的に,先述の一般的な拡散係数を示すMOF中のCO2に対しては,室温付近およびそれ以上の温度下ではCP法が効かない。まれに,細孔内部の立体障害や吸着サイトとの強い相互作用などによってCO2の拡散速度が遅く,CP法でCO2の13C信号を観測できる場合がある30–33)。拡散速度を調べる測定法ではないため,拡散NMRのように拡散係数を求めることはできないが,拡散係数が一般的な値程度かそれより小さいかの判断など,定性的な解析は可能である。
2.4 同位体核種濃縮CO2ガスの使用
13C核の天然存在比は1%程度であり,残りの99%はすべてNMRで観測できない12C核である。そのため,特にCO2の吸着量が少ない圧力条件下や,信号が広幅な静止状態では,信号を観測するためのスペクトルの積算に数時間から場合によっては1日以上を要することが予想される。信号の感度向上のため,費用はかかるが13C同位体濃縮したCO2ガスを利用することが望ましい。なお,17O同位体濃縮ガスを用いれば固体17O NMRによる測定・解析も可能であり,そのような例も報告されている34)。本稿では解説しないが,17O核は四極子相互作用も持つため,化学シフト相互作用と組み合わせることでより詳細な運動状態の解析が可能となる。
2.5 CO2雰囲気下測定のための試料管準備
CO2を吸着した試料の固体NMR測定は,試料管内に試料とCO2ガスを入れた状態で行われる。一般的な固体NMRの回転試料管は気密性に乏しいため,測定には何らかの工夫が必要となる。気密性を高めた特殊な試料管のキャップも売られており,例えばCO2を充填したグローブボックス中などで試料管に取り付けることで,試料管内に試料とCO2を閉じ込めることができる。しかしこの場合,ガスの使用量が多いため同位体濃縮ガスは使用できず,また圧力も大気圧に限られる。そこで本節では,CO2雰囲気下で測定を行うための試料管準備の方法を紹介する。
2.5.1 ガラス管の使用
CO2を含む特殊雰囲気下における固体NMR測定で一般的に行われるのが,試料を詰め任意の圧力のガスを充填したガラス管をガスバーナーで焼き切って封じる方法である。MOFにおいては,吸着したCO2の固体NMR測定のほとんどがこの方法で行われている。ガラス管の封管には慣れが必要だが,静止試料の測定はこれで問題なく行うことができる。ただし,MASを行いたい場合,封管したガラス管が回転試料管に入るため,回転による遠心力でガラス管が破損するリスクや,ガラス管の形状によっては回転が不安定になり試料管が破損するリスクがある。
NMR装置外部でガスボンベに接続した単結晶サファイア製の試料管をNMRプローブ中に設置することで,測定環境下で試料に様々なガス圧を印加するin situ測定を行った例が報告されている35, 36)。このような装置は筆者が知る限り市販されておらず,自作が必要となるが,一つの試料に対して連続的にガス圧を変化できる点は大きなメリットである。先行研究においてはCO2を大気圧以下~約10気圧,Xeでは20気圧近くまで変化させ,固体13Cおよび129Xe NMR測定を行っている。ガラス管で同様の装置を作製することで,サファイアと比べて耐圧性は低下するものの,様々なガス圧力下で測定を行う場合には有効に運用できると期待される。ガラス管で作製した装置でex situ測定を行った例も報告されている37)。
2.5.2 特殊な回転試料管の作製
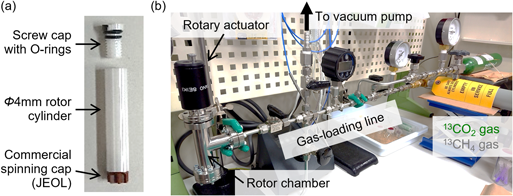
市販の回転試料管の規格を基に,Oリングの付いたスクリュー式キャップを備えた高気密性の回転試料管を設計した報告がある38)。筆者らはこの報告を参考に,図4に示すような気密性回転試料管を設計・作製し,更に任意の圧力のガス雰囲気下で試料管のキャップを開閉できるガス配管を開発することで,CO2雰囲気下におけるMOFの13C MAS NMR測定を行っている33, 39)。これまでに10気圧程度までは漏れなくCO2を封入できており,ガラス管と比べてMASによる高感度・高分解能測定が容易である点や,1度試料管に詰めた試料に対し様々なガス圧を印加できる点などから,利便性の高い手法と言える。
MOFは,金属イオンと配位子の配位結合から形成される多次元的な骨格を持った多孔性材料である。用いる金属種と配位子の組み合わせによって多彩な構造が合成でき,また配位子への置換基の導入などによって細孔中に立体障害や新たな吸着サイトを導入可能といった特徴を持つ。こうした構造の設計性の高さから,これまで数万種類のMOFが作製され,その吸着特性が盛んに研究されている。固体NMRはMOFの局所構造やゲスト分子の吸着状態の解析にしばしば用いられてきたが,吸着したCO2のダイナミクスが固体13C NMRによって解析されるようになったのはここ10年ほどであり,報告されている文献も筆者が知る限り30報程度とそれほど多くはない。しかしながら,その多くが固体NMRによる解析特有のCO2の吸着状態の情報を得ることに成功している。以下では,代表的ないくつかの論文について取り上げ,解説する。
3.1 CO2のローカル運動
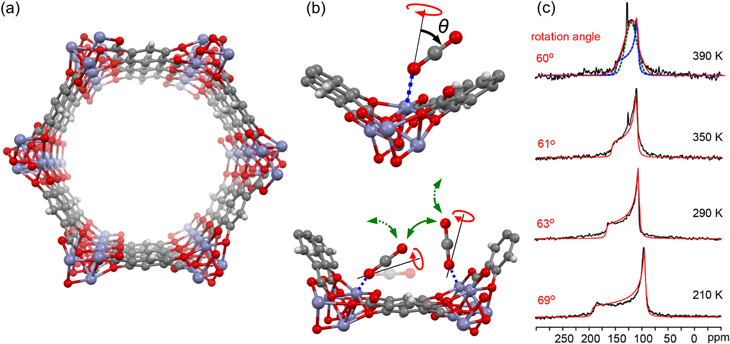
固体13C NMRによってMOFに吸着したCO2の運動性を解析する試みは,2012年にU. C. BerkeleyのReimerグループにより初めて行われた40)。用いられたのはMOF-74と呼ばれるハニカム型構造の1次元細孔を有するMOF(図5a)であり,化学的安定性の高さなどから盛んに研究されている。ReimerらはMOF-74中のCO2に対し,13C CSAによる粉末パターンを解析することで,吸着状態のCO2が一軸回転運動を行うことを初めて提唱した。回転運動の傾きの角度θは~60–70°程度の範囲であり,温度が上昇するとθが小さくなることや,また吸着量によってもθが変化することを示した(図5b,c)。また,13C T1緩和時間の温度変化の解析よりEaを求め,吸着量増加によりEaが小さくなることからCO2と吸着サイトの相互作用が弱まっている可能性を指摘した。このように,固体NMRでCO2の吸着状態を詳細に解析できることを示した重要な研究である。彼らはその1年後,モンテカルロ法による細孔中のCO2の自由エネルギーの計算より,MOF-74中のCO2は一軸回転運動でなく揺動運動と吸着サイト間のホッピング運動を行っている可能性を提案し,そのような2種類の運動の組み合わせでもCSAの粉末パターンを再現しうることを報告している41)。その翌年にはThe University of Western OntarioのHuangらによって固体17O NMR測定が行われ,17O核のCSAと四極子相互作用が合わさった線形の解析により,θ~20–30°程度の一軸回転運動と吸着サイト間のホッピング運動(図5b)が起きていると提案された34)。
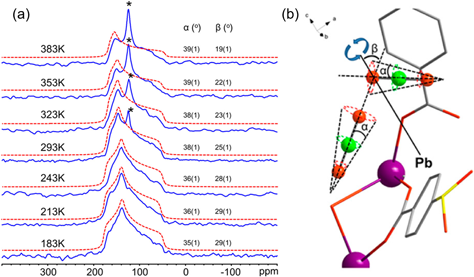
HuangグループはMOF-74以外にも,様々なMOF中に吸着したCO2のダイナミクス解析に取り組んでいる。彼らは13C CSAによる粉末パターンを利用したローカル運動の解析に加え,13C CP測定やXRD,計算科学手法などによる静的な吸着構造の解析も行うことで,CO2の吸着状態を効果的に解析している。例えば,CdSDBおよびPbSDBと呼ばれるCd2+およびPb2+からなる構造のMOFに対し,単結晶XRDと固体13C NMRを組み合わせた解析を行っている42)。CO2吸着下での単結晶構造解析によってMOF中のCO2吸着位置を特定し,また13C粉末パターンを解析することで,CO2が吸着サイト上でどのように運動し,また吸着サイト間をどのようにホッピングするのかを明らかにした(図6)。固体NMRは局所的な静的・動的構造の解析を得意とする一方,結晶構造のような全体構造を把握することは難しく,またXRDは逆の性質を持つため,これら二つの手法を相補的に組み合わせることは非常に有効と言える。α-Mg formateと呼ばれるMg2+とギ酸イオンからなるMOF中のCO2に対しては,13C粉末パターンによる一軸回転運動およびホッピング運動の解析に加え,分子動力学計算によって細孔中のCO2の位置を解析している43)。計算で得られたCO2の位置の分布はNMRから得られた分子運動の様子とよく一致しており,計算結果の妥当性を実験的に示しつつ,細孔内でのCO2の動きや吸着部位を可視化することに成功している。
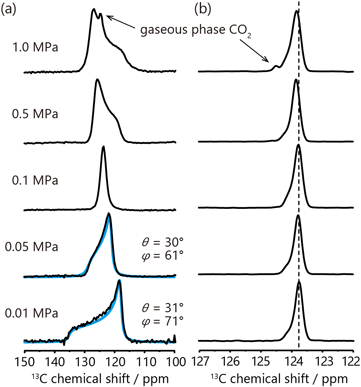
上記の例はすべて静止状態の試料の固体NMR測定により行われているが,MAS法を用いた解析の例も紹介する。UTSA-16と呼ばれるMOFに吸着したCO2に対し,MASによりCSAが平均化され先鋭化した13C信号のピーク位置の温度変化から,温度に応じた吸着部位の変化が解析されている22)。ピーク位置の変化の幅は1 ppm程度であるが,MASを行わない場合はCSAとMOF中の常磁性Co2+イオンの影響で粉末パターンは300 ppmに及ぶ線幅を示しており,このような等方化学シフト値の小さな変化が検出できたのはMAS法を用いたためと言える。筆者らは気密性の回転試料管を作製し,先述のMOF-74に対し0.01–1 MPaのCO2圧力下で13C MAS NMR測定を行った39)。MASで先鋭化したCO2の13C信号からは圧力上昇に伴う0.1 ppm程度のピークシフトが観測されている(図7)。MOF-74中においてCO2はガス圧の上昇に伴い,低圧時に吸着する第1吸着サイトとは異なる第2吸着サイトにも吸着し,それらサイト間のホッピングによる交換の様子がピークシフトとして現れていると解釈できる44)。10–20 ppm程度の線幅を示す静止状態の粉末パターンの解析では,0.1 ppm程度のピークシフトはノイズによる誤差に埋もれてしまう可能性があり,やはり等方化学シフト値の解析におけるMAS法の有効性がうかがえる。
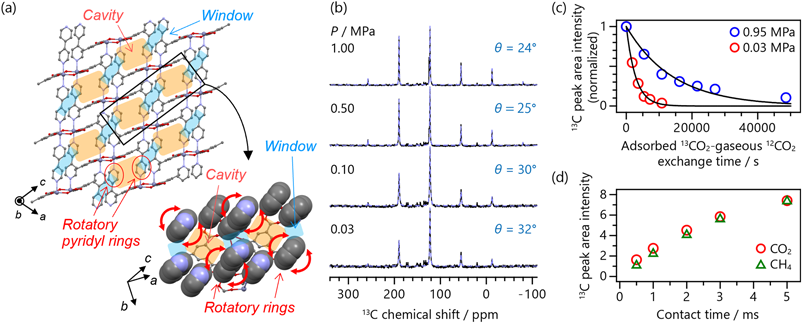
CO2の一軸回転運動の傾きおよびホッピング運動による角度変化が小さいとき,粉末パターンは線幅が200–300 ppmに及ぶような非常に広幅な信号を示す。このような場合は静止状態で測定するより,数kHz程度でMASを行いスピニングサイドバンドによって信号をスパイク上にすることで,測定感度を大きく向上しつつCSA解析を行える。筆者らは,CID-Meと呼ばれるMOF(図8a)の狭い1次元細孔中に吸着したCO2に対し,13C MAS NMR測定を行いその運動性を解析した33)。細孔中の吸着部位であるキャビティ部にCO2が1分子吸着する0.03 MPaから2分子吸着する1 MPaまで圧力を変化させて測定し,サイドバンドパターンに対するフィッティングでCSA値を取得し式(2)によって回転の傾きθを求めた(図8b)。その結果,0.03 MPaではθ=32°,1 MPaではθ=24°と吸着量の増加に伴うθの減少が確認された。これは,CO2が狭い細孔内にて配位子や別のCO2による立体障害を受け,運動が制限されていることを示している。CID-Meは細孔のウィンドウ部(図8a参照)において配位子の一部が回転運動を有しており,この回転の際に細孔径が2.5 Åから3.4 Å程度まで動的に拡大する特徴を持つ。筆者らは固体2H NMR測定により,高圧下ではキャビティ部に2分子吸着したCO2が立体障害となり,配位子の運動性も制限される様子を観測した。これらの知見を基に,配位子の運動性の低下による細孔径の拡大の抑制がCO2の細孔内拡散に影響を及ぼすことを見出しており,その詳細は次節にて紹介する。
3.2 CO2の細孔内拡散
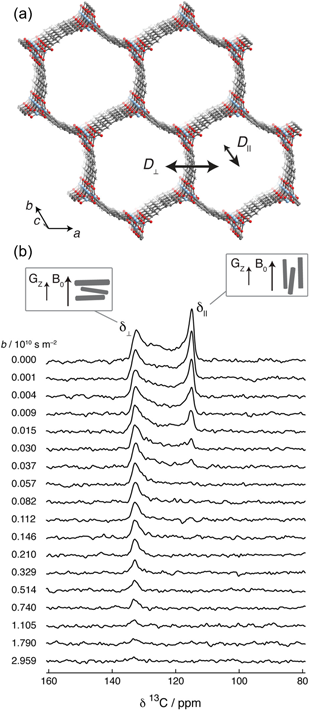
CO2の拡散の解析では,多くの場合拡散NMR法が用いられている。MOF細孔中におけるCO2に関しては,先述の通り,10−8–10−10 m2/s程度の拡散係数が拡散NMR測定により観測されている。通常の測定では単一の拡散係数が得られるのみにとどまるが,中には粉末パターンを利用することで,細孔構造に依存した拡散速度の異方性を解析した報告もある。2.1節で述べたように,固体試料は異方性相互作用のため,NMRの静磁場に対する結晶や分子の向きによってピーク位置が変化し,その足し合わせが粉末パターンである。吸着したCO2の場合も同様に,ホストである結晶の磁場に対する向きとともにCO2の向きが変わり,CSAにより13Cピークの位置が変化する。ここで,例えば結晶のa軸方向とbc軸方向で細孔径が異なるような異方的な細孔構造の場合,結晶の向きとともに異方的な細孔の向きも変化するため,CO2の13C粉末パターンは磁場に対する細孔の向きを反映したものとなる。これに対して拡散NMR測定を行い,磁場勾配による粉末パターンの変化を解析することで,拡散係数の細孔方向依存性を測定した例がいくつか報告されている27–29, 37)。ここでは一例として,先述の1次元細孔を持つMOF-74の同形構造体(図9a)に対する,Reimerらによる測定結果を紹介する28, 37)。実験では1次元細孔方向に長い針状の結晶が使用され,図9bに示すように,吸着したCO2の粉末パターンの低周波数側および高周波数側のエッジが,それぞれ静磁場B0方向に対し結晶が平行および垂直に向く場合と対応する。磁場勾配パルスGzをB0と平行方向に印加しながら13C拡散NMR測定を行ったところ,パルス強度の増加に伴い粉末パターンの低周波数側がより速く強度が減衰する様子が観測された。このことは,1次元細孔方向の拡散の方が,細孔と垂直方向の拡散よりも速いことを示している。彼らはスペクトルシミュレーションによる解析より,1次元細孔方向の拡散係数(D//)を6.5×10−9 m2 s−1,細孔と垂直方向(D⊥)を3.9×10−11 m2 s−1と得ている。1次元細孔にも関わらず細孔と垂直方向にCO2が拡散できるのは,欠陥により細孔壁に穴が空いているためとされる。
筆者らは拡散NMR以外の手法で拡散速度を見積もる試みを行っており,これについても紹介する。3.1節で述べたように,筆者らはCID-Meと呼ばれるMOFの1次元細孔中にて,CO2の吸着に伴い配位子の運動性が低下する様子を観測した。このCID-Meの細孔径は配位子の回転の際に拡大する性質を持ち,したがって高圧下にて回転速度が低下することで細孔径拡大が抑制され,CO2の拡散が遅くなると予想される。これを実証するため,初めにMOFに13C同位体濃縮CO2(13C 99%)を吸着させ,その後MOFを通常のCO2(12C 99%)下に置くことで細孔内の13CO2と12CO2を交換し,その速度を13C MAS NMRでモニターするという実験を行った。細孔内に吸着したCO2が細孔外の気体のCO2と入れ替わる過程では細孔内の拡散を経ることになるため,交換速度には拡散速度が反映されると期待される。圧力条件を細孔キャビティ部にCO2が一つ吸着する0.03 MPaおよび二つ吸着する0.95 MPaと設定し,図4に示したスクリューキャップ式の気密回転試料管および専用に設計した配管を用いることで各圧力下において13CO2を吸着したMOFを12CO2に晒し,13C信号の経時変化を観測した。その結果,図8cに示すように吸着した13CO2の信号強度が12CO2との交換で徐々に減衰していく様子が確認された。指数関数によるフィッティングより交換速度は0.03 MPaでは3.5×10−4 s−1,0.95 MPaでは7.0×10−5 s−1と得られ,配位子の回転速度が低下する高圧下にてCO2の細孔内拡散速度も低下することが示された。なお,実験に用いたMOFの結晶サイズ(~10–50 µm)より拡散係数を概算すると10−13–10−15 m2 s−1となり,一般的なMOF中のCO2の拡散よりもだいぶ遅いことがわかる。これは狭い細孔中における回転する配位子の立体障害の影響であり,このような細孔構造だからこそ配位子の回転速度がCO2の拡散に影響を及ぼしたと考えられる。
このCID-Meの研究において,筆者らはCP法を用いた拡散速度の解析も行った。CID-Meに吸着したCO2は遅い拡散によりCP法で13C信号が観測できる。CPにおける磁化移動の速度は,核間距離に依存した双極子相互作用の大きさによる。そのため,磁化を移すCPパルスの照射時間(コンタクトタイム)に依存してどのように磁化が増幅されるかを調べることで,核間距離の情報が解析できる。研究ではCO2と比較的分子サイズが近く,また一般にCO2より吸着が弱いCH4について,CID-Meに吸着した場合CP法で13C信号が観測できることを確認したため,その信号強度のコンタクトタイム依存性を測定し,CO2との比較を行った。その結果,図8dに示すように両分子ともほぼ同様のコンタクトタイム依存性を示した。CH4は吸着状態でも分子全体が回転運動をしており45),これにより分子内の1H–13C双極子相互作用が平均化されCPに寄与しないため,CO2同様に配位子の1Hからの磁化移動で信号が観測されている。したがってこの結果は,CO2およびCH4の13C核と配位子の1H核との平均核間距離が同程度,すなわち同じような速度で拡散していることを示している。一般に吸着の弱いCH4の方が細孔内を拡散しやすいと予想されるが,狭い細孔内では吸着の強さよりも立体障害が拡散速度に効くと考えられる。このような状況下で立体障害となる配位子の回転速度を制御できれば,ガスの拡散速度は分子サイズ選択的にコントロール可能であることを本研究は提案する。