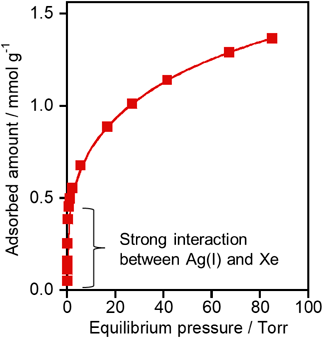
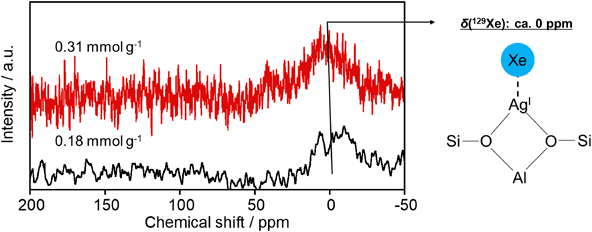
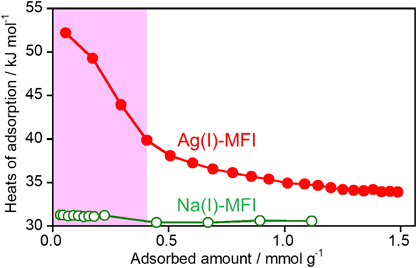
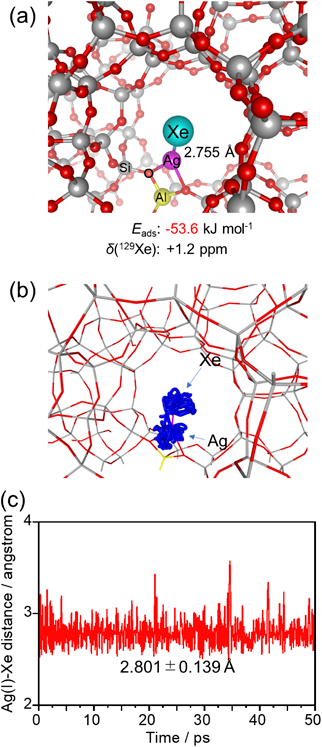
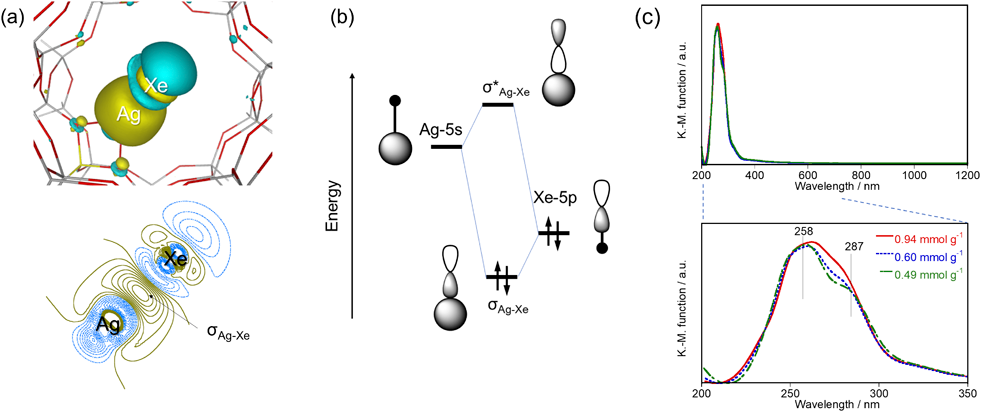
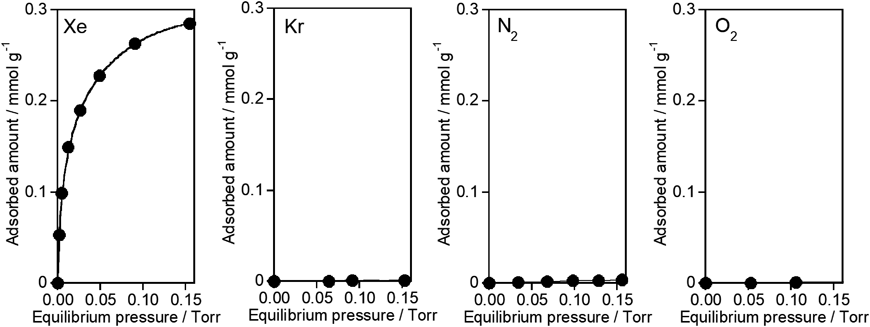
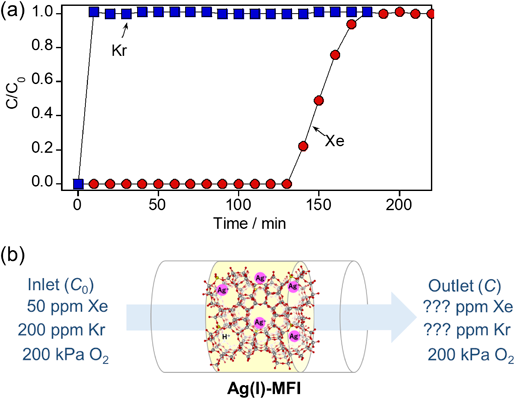
ゼオライト細孔内で観測されたAg(I)–Xe化合物形成を駆動力とする室温低圧下におけるXe選択捕集Selective Collection of Xe under Low Pressure at Room Temperature Driven by the Formation of Ag(I)–Xe Compound Observed in MFI Zeolite Pores
1 名古屋大学大学院工学研究科Department of Materials Chemistry, Graduate School of Engineering, Nagoya University ◇ 〒464–8603 愛知県名古屋市千種区不老町
2 岡山大学学術研究院自然科学学域Department of Chemistry, Graduate School of Natural Science and Technology, Okayama University ◇ 〒700–8530 岡山県岡山市北区津島中3–1–1
受理日:2022年9月11日Accepted: September 11, 2022
発行日:2023年1月31日Published: January 31, 2023