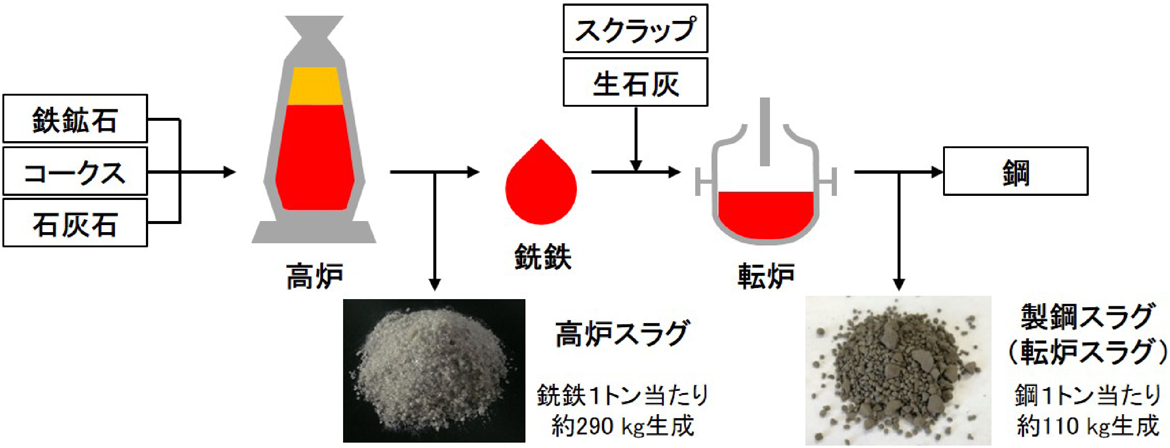
“スラグ”は鉱石から金属を製錬したときに生じる副産物であるが,最も多く生産されているのが鉄鉱石から鉄鋼製品を製造する還元・製錬工程で発生する鉄鋼スラグである。鉄鋼スラグは,高炉スラグと製鋼スラグの二種類に大別される。前者は鉄鉱石を石灰石やコークスとともに高炉で溶融・還元し銑鉄を製造する製銑工程で副生成し,後者は製鋼炉で溶鋼中の不純物を除去して鋼を製造する製鋼工程で副生成する(Fig. 1)1)。2019年度における鉄鋼スラグの国内生産量は3617万トンにも及ぶ(高炉スラグ2275万トン,製鋼スラグ1342万トン)1)。国内の火力発電所で副生成される石炭灰の生産量が年間900万トン程度であることを考えると,いかにその生産量が多いかがわかる。鉄鋼スラグはそのままでは処理費用が嵩むため,水硬セメント,コンクリート骨材,路盤材,舗装材などといった土木建築用材料の原料として,現在そのほとんどが再利用されているが,公共事業の縮小化や国内スラグ処分場の確保,環境規制の厳格化などといった問題から新たな利用法の開拓が求められている2,3)。
高炉スラグは,鉄鉱石に含まれる鉱物成分と製錬過程で融剤として加えられる石灰石とが溶融・結合してできるため,主成分としてCaO(約40%),SiO2(約35%),Al2O3(約15%),MgO(約5%)の他,FeO,MnO,TiO2,Sなどの微量元素を含んでいる。一方,製鋼スラグは銑鉄を鋼へと精錬する各工程において様々な化学組成のものが生成するが,80%以上をCaO(約30 – 55%),FeO(約14 – 40%),SiO2(約9 – 19%)が占め,残りをその他の酸化物(MgO, MnO, Al2O3, P2O5)が占める1)。いずれのスラグも,これらの元素が酸素原子を介して複雑に絡み合い,後処理によっては結晶相を包含した構造を持つ。鉄鋼スラグの組成はこのように複雑ではあるが特徴ある化学組成を有している。例えば,SiO2(およびAl2O3)はゼオライトやメソポーラスシリカなどのシリカ多孔体の構成成分である。鉄鋼スラグを原料にしてこれらの材料を合成できれば,珪酸ソーダやシリカアルコキシドなどを原料とする従来の合成法よりも安価かつサステイナブルなシリカ多孔体製造プロセスとなる可能性がある。また,鉄鋼スラグに最も多く含有されるCaO成分は,高い固体塩基性を示すだけでなく,700°C付近の温度域で可逆的な炭酸化/脱炭酸反応(CaO(s)+CO2(g)=CaCO3(s))を生じ,CO2吸着材としての機能を示す。さらに,鉄鋼スラグに含まれるMnやFeなどの遷移金属元素は触媒活性金属として特定の反応に利用できる可能性がある。すなわち,鉄鋼スラグは,高付加価値な機能性材料合成のための安価な化学原料としてのポテンシャルを有している4,5)。しかしながら,多数の元素が強固な金属–酸素結合を介して複雑に絡み合った独特の化学組成を有する鉄鋼スラグから,これらの有価な金属を選択的に抽出することは容易ではない。
筆者らはこれまでに,簡便な化学プロセスを用いて鉄鋼スラグに含まれる成分を溶出・再構築することで,ゼオライト–ハイドロキシアパタイト複合体6,7)やCaO–メソポーラスシリカ複合体8,9),高表面積カルシウムシリケート10),Ca系層状複水酸化物11)へと変換する手法を開発してきた。いずれの手法も,合成に当たっては安価な酸および塩基性水溶液を用い,合成温度100°C以下,濾過や蒸発乾固といった簡便な工程からなり,大量の廃スラグを処理するためのラージスケール合成にも適したプロセスである。また,鉄鋼スラグを原料に合成したこれら材料群が,試薬から合成した純正品とほぼ同等の吸着能・触媒能を示すことを実証してきた12–16)。本稿では,このうち,ゼオライト–ハイドロキシアパタイト複合体およびCaO –メソポーラスシリカ複合体への変換プロセスについて,その概要と合成方法および合成した材料の吸着材としての応用について解説する。
2. 高炉スラグを原料としたハイドロキシアパタイト–ゼオライト複合体の合成
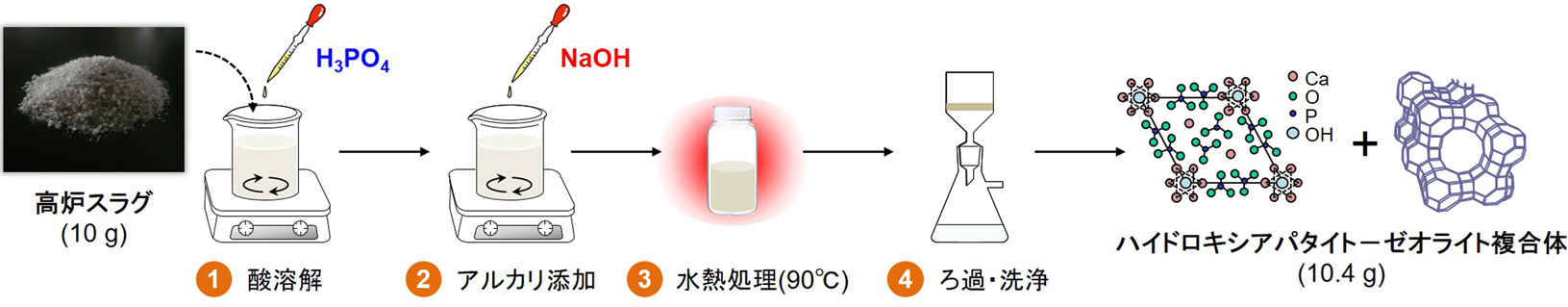
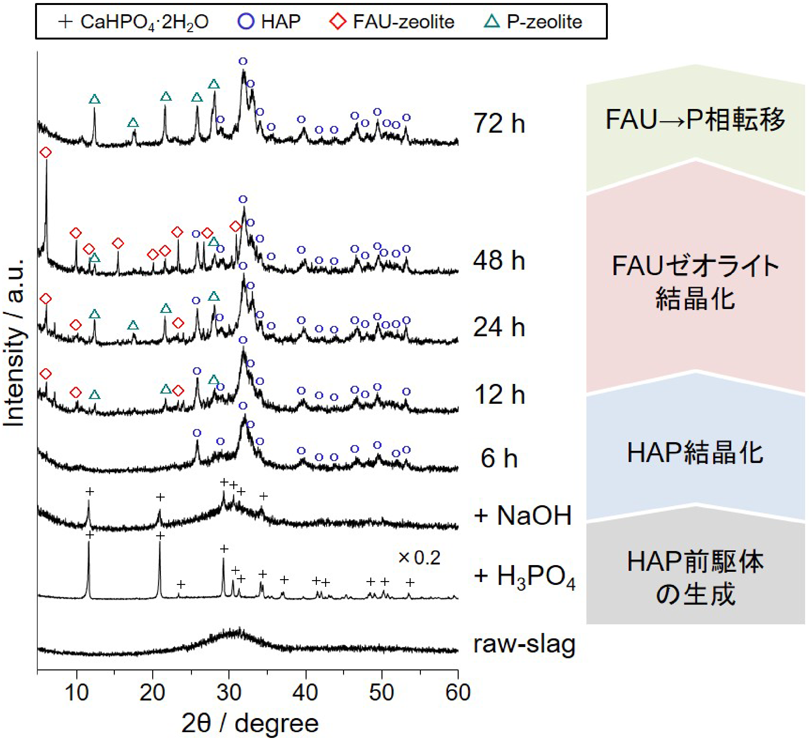
高炉スラグに含まれる豊富なSiO2,Al2O3成分に着目し,ゼオライトへと変換する試みは従来から行われていた17,18)。例えば,フライアッシュやもみ殻灰のようなSiO2を主成分とする高融点複合酸化物を原料にゼオライトを合成する際には,①酸または塩基を用いた溶解による金属成分の抽出,②Al原料の添加によるSi/Al比の調整,③水熱処理によるゼオライト結晶の析出を経るプロセスが定石である19)。しかしながら,高炉スラグにはCaO(およびMgO)成分が多量に含まれており,これがCa2+(およびMg2+)イオンとして母液に混入しゼオライトの核生成を妨げるため,上記の方法では合成することができない。そのため,塩酸や硝酸を用いて高炉スラグから金属成分を抽出した後,加熱することでシリカゲルを生成させ,濾別することでSiO2成分を分離し,これをゼオライト合成の原料に用いていた。しかしこの場合,スラグ含有成分のうち利用できる金属成分は35%程度であり,Ca,Mg,Alなどの他の成分の多くは除去されてしまうため,効率的なスラグ再利用プロセスとは言い難い。また,煩雑な合成プロセスには常にエネルギーコストの問題が介在すると同時に,廃スラグの大量処理には適さない。これに対し筆者らは,Caと反応性の高いリン酸を用いることにより,一段階でハイドロキシアパタイト–ゼオライト複合体(HAP-ZE)を合成するプロセスを開発した(Fig. 2)6,7)。本プロセスは,①高炉スラグをリン酸で溶解することにより金属成分を抽出すると同時に,ハイドロキシアパタイト前駆体を生成させる工程,②水酸化ナトリウムを加えて溶液pHをアルカリ条件にし,ハイドロキシアパタイト(HAP)を結晶化させる工程,③溶液を90 – 100°Cで熟成し,ゼオライトを結晶化させる工程,④固体生成物を濾過,洗浄,乾燥する工程からなる。すなわち,まずゼオライトの核生成を阻害するCa成分をリン酸カルシウム化合物として析出させることで除去してしまおうという発想である。この際加えるリン酸と水酸化ナトリウムの量は,それぞれHAPの化学量論比Ca/P=1.67,およびフォージャサイト(FAU)型ゼオライトの最適な合成条件Na/Al=10となるように調整した。Fig. 3には90°Cでの熟成時間に伴う生成物のX線回折(XRD)パターンの変化を示す。原料の高炉スラグはアモルファス構造を有する砂状の粒子であり,比表面積は8 m2/g程度である。高炉スラグをリン酸(1 mol/L)で溶解することで,まずHAP前駆体であるリン酸水素カルシウム水和物(CaHPO4·2H2O)が生成した。ここに水酸化ナトリウム水溶液(3 mol/L)を加えることで,pHの上昇に伴って12 h程度でHAPの結晶化が進行し,その後逐次的にFAUゼオライトが生成,熟成48 hで最も結晶性の高いFAUゼオライトが得られた。このような逐次的結晶化過程は,HAP生成によって溶液中のCa2+およびMg2+イオン濃度が十分低下した後にゼオライトの核生成が誘発されたためであると考えられる。また,長時間熟成させるとFAUゼオライトはより緻密な結晶相を有するPゼオライトへと相転移した。なお,リン酸の代わりに塩酸や硝酸を用いた場合にはCa2+イオンが析出せず母液中に存在し続けるため,ゼオライトは形成されなかった。このことから,高炉スラグからのゼオライト合成においてリン酸が果たす役割がいかに重要かわかる。
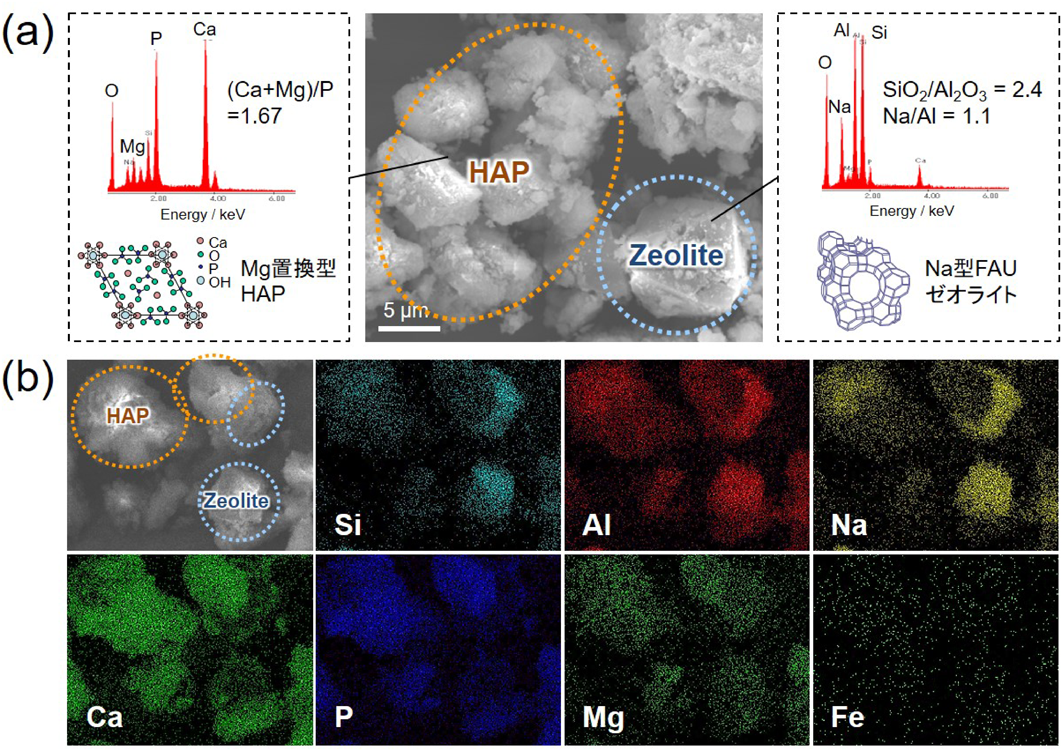
熟成時間を48 hとして合成した試料には,無定形のHAP粒子と特有の八面体構造を有するFAUゼオライト粒子が観察された(Fig. 4(a))。エネルギー分散型X線分光(EDX)により各粒子の化学組成を分析したところ,HAP粒子からは主にCa,Mg,P,Oが検出され,(Ca+Mg)/P比はHAPの化学量論比である1.67に近いことから,Mg置換型HAPであると推察された。ゼオライト粒子からは主にSi,Al,Na,Oが検出され,Na/Al比はおよそ1.1,SiO2/Al2O3比はおよそ2.4であることから,Na型FAUゼオライトの形成が確認された。すなわち,本プロセスにおいてリン酸はスラグを溶解させる溶解酸としての役割とHAPの化学原料としての役割を,また,水酸化ナトリウムはpHを塩基性にするアルカリとしての役割とゼオライトの金属カチオン原料としての役割を果たしていることがわかる。また,FeやMnなどの微量元素は両粒子中に分散して存在しており,ゼオライトの結晶成長を阻害することなく最終生成物中に取り込まれていることがわかった(Fig. 4(b))。N2吸脱着測定から,得られた複合体はゼオライトに特徴的なI型の吸着等温線を示し,BET比表面積は194 m2/gであり,0.8 nm程度のマイクロ孔を有することも確認された。化学分析から求めた複合体に含まれるHAPとゼオライトの重量比がおよそ68 : 32(wt%)であり,標準的なFAUゼオライトの比表面積が600 – 700 m2/g程度であることを考えると,十分結晶化度の高いFAU粒子が形成されているものと考えられる。
このような化学プロセスにおいて問題となるのは,原料である高炉スラグの化学組成の均質性である。高炉スラグの化学組成は各製鉄所により若干のばらつきはあるが,本プロセスにおいては上述した出発原料比となるようにリン酸の量を調整すれば安定してハイドロキシアパタイトとゼオライトの複合体を得ることが可能である。高炉スラグに含まれるSiとAlの比率(本研究で用いた高炉スラグではSi/Al=2.0)にも若干のばらつきはあるが,構造規定剤フリー条件下でのFAUゼオライト合成における出発ゲルSi/Al比は2から3程度まで許容されるため,組成が多少異なってもFAUゼオライトが得られる可能性が高い。また,本合成プロセスは不要成分の除去や分離を必要としないため,スラグ成分の再利用効率が高いことが利点である。実際,本プロセスでは高炉スラグ10 gから10.4 gのHAP-ZEを得ることができた。これは,金属原子数基準で高炉スラグに含まれる金属成分の実に85%以上を再利用できた計算になる。
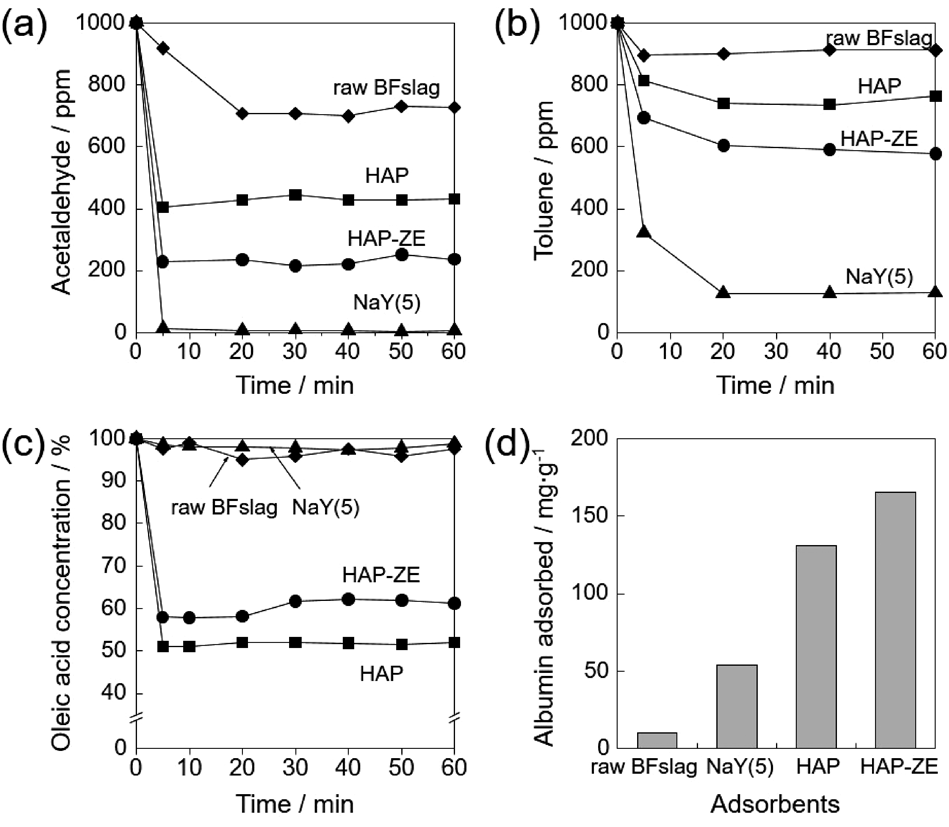
生成物はHAPとゼオライトとの混合物として得られ,また,スラグ由来不純物元素を含むため,高い選択性や純度が要求される場面での利用には適さない。このため,“質”よりも“量”が必要とされる環境保全分野での吸着材としての応用を検討した。ゼオライトは水や揮発性有機物の吸着特性に優れ,HAPは脂肪酸や蛋白質に対し優れた吸着特性を有する。高炉スラグから合成したHAP-ZEの吸着特性について,空気中のアセトアルデヒドおよびトルエン(各1000 ppm)の吸着実験により評価したところ,原料の高炉スラグ(BFslag)はほとんど吸着能を示さないが,HAP-ZEは市販のNaY(SiO2/Al2O3=5)ゼオライトのおよそ45%の吸着量を示した(Fig. 5(a), (b))。また,オレイン酸(n-hexane溶媒中2.4 mmol/L)の吸着実験により評価したところ,HAP-ZEは市販のHAPのおよそ80%のオレイン酸吸着量を示した(Fig. 5(c))。これらの吸着量はHAP-ZEに含まれるゼオライトおよびHAPの構成比とほぼ一致しており,それぞれ,純粋なゼオライトおよび純粋なHAPに匹敵する吸着能を持つことがわかった。また,水中に分散した蛋白質に対しても吸着能を示した(Fig. 5(d))。ゼオライトの陽イオン交換能とHAPの陽・陰イオン交換能を利用すれば,水中の重金属イオンや放射性同位体元素(137Cs,90Srなど)の吸着材としての利用の可能性もあり,現在我々も検討を行っている20)。本プロセスを利用すれば,高炉スラグを出発原料として汎用性の高い吸着材を安価に製造でき,大量供給にも対応可能な環境保全用吸着材製造プロセスになりうると考えている。
3. 高炉スラグを原料としたCaO–メソポーラスシリカ複合体の合成とCO2吸着への応用
現在,鉄鋼産業が直面しているもう一つの重要な問題が,高炉で還元剤として添加されるコークスや融剤として添加される石灰石(CaCO3)の使用により発生する大量の二酸化炭素(CO2)の排出である。CO2は平成29年度において国内で年間11億9000万トン排出されており,このうち鉄鋼産業から排出されるCO2は年間1億6300万トンに上り,国内CO2総排出量の約14%に相当する21)。2008年から始まった革新的製鉄プロセス技術開発(COURSE50)でも2050年までに鉄鋼産業におけるCO2排出量の約30%削減が目標に掲げられており,鉄鋼産業におけるCO2回収貯留(CCS: Carbon Capture and Storage)は喫緊の課題となっている22,23)。このような社会的要請から,アミン系水溶液を使用した排ガスからのCO2吸着が有望なプロセスとして開発・利用されているが,アミン溶液の再生にかかるエネルギーや,アミン溶液自体の劣化,設備腐食などの運用上の問題があることが主な欠点である24,25)。高炉や石炭火力発電所で発生する排ガスからのCO2回収の代替法として,固体であるCaOの可逆的炭酸化/脱炭酸反応(CaO(s)+CO2(g)=CaCO3(s), ΔH°298=−178.2 kJ/mol)を利用する方法(カルシウムルーピング)がある26–28)。CaOは高い理論CO2吸着量(0.78 g−CO2/g−CaO)を示すことに加え製造コストが安価なことから,セメント産業などではセメント製造時に副生する水酸化カルシウム(Ca(OH)2)を利用したCO2吸着技術が既に実用化されている。しかしながら,炭酸化/脱炭酸反応に700°C付近の高温を必要とするため,稼働時のエネルギーコストや高温での連続使用による吸着材の劣化が課題となっている。このため,CaOを他の安定な酸化物担体(SiO2,Al2O3,Fe3O4など)に固定化して吸着材の耐久性向上を図るなどの対策が検討されている29,30)。
高炉スラグの成分のうち大部分を占めるCaO(およびMgO)は,可逆的炭酸化/脱炭酸反応によりCO2吸着に利用できる成分である。ただし,高炉スラグのような複合酸化物のマトリクス中に固溶したCaはCO2吸着能を持たず,CO2吸着能を発現させるためには結晶相としてのCaOを形成させる必要がある。また,残りの成分の多くを占めるSiO2成分は規則配列したメソ細孔と高い比表面積を有するメソポーラスシリカ(MS)の構成元素である。そこで筆者らは,化学的アプローチによって高炉スラグをCaOとメソポーラスシリカの複合体へと直接変換できれば,有用なCO2吸着材として利用できるのではないかと着想した。
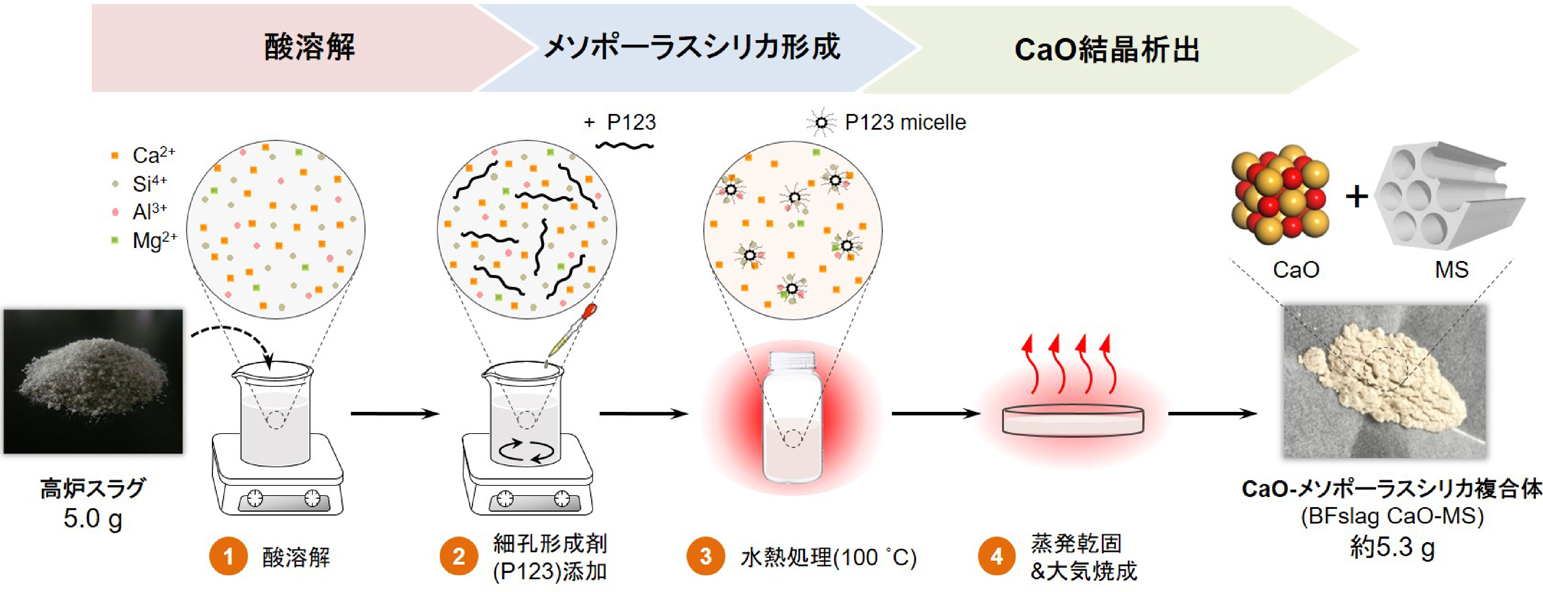
高炉スラグを原料としたCaO –メソポーラスシリカ複合体(BFslag CaO-MS)の合成は,SBA-15メソポーラスシリカの合成法31)を改良して行った8)。まず,高炉スラグに3.0 mol/Lの酸を加え,完全に溶解させて高炉スラグに含まれる金属イオンを抽出した(Fig. 6,ステップ1)。このとき酸として三種類の酸(ギ酸(FA),硝酸(HNO3)または塩酸(HCl))を使用した。この酸溶液に,細孔形成剤であるPluronic P123®を加え,このときの各物質のモル比がP123:Si(高炉スラグに含まれるSi):酸:H2O=0.017 : 1 : 5.7 : 193となるように調整した(ステップ2)。こうして得られた溶液を40°Cで24時間熟成した後,テフロンボトルに移し100°Cの静置条件下でさらに24時間水熱処理した。この段階で,Si4+イオンがP123ミセルを取り込みながら縮合重合し,メソポーラスシリカが形成される(ステップ3)。得られた懸濁液からエバポレーターを用いて水を蒸発乾固させ,大気中600°Cで焼成した(ステップ4)。この段階で溶液中に残存していたCa2+イオンが析出することでCaO結晶が形成される。ギ酸を溶解剤として使用した場合,5.0 gの高炉スラグから約5.3 gのBFslag CaO-MS複合体が合成できた。
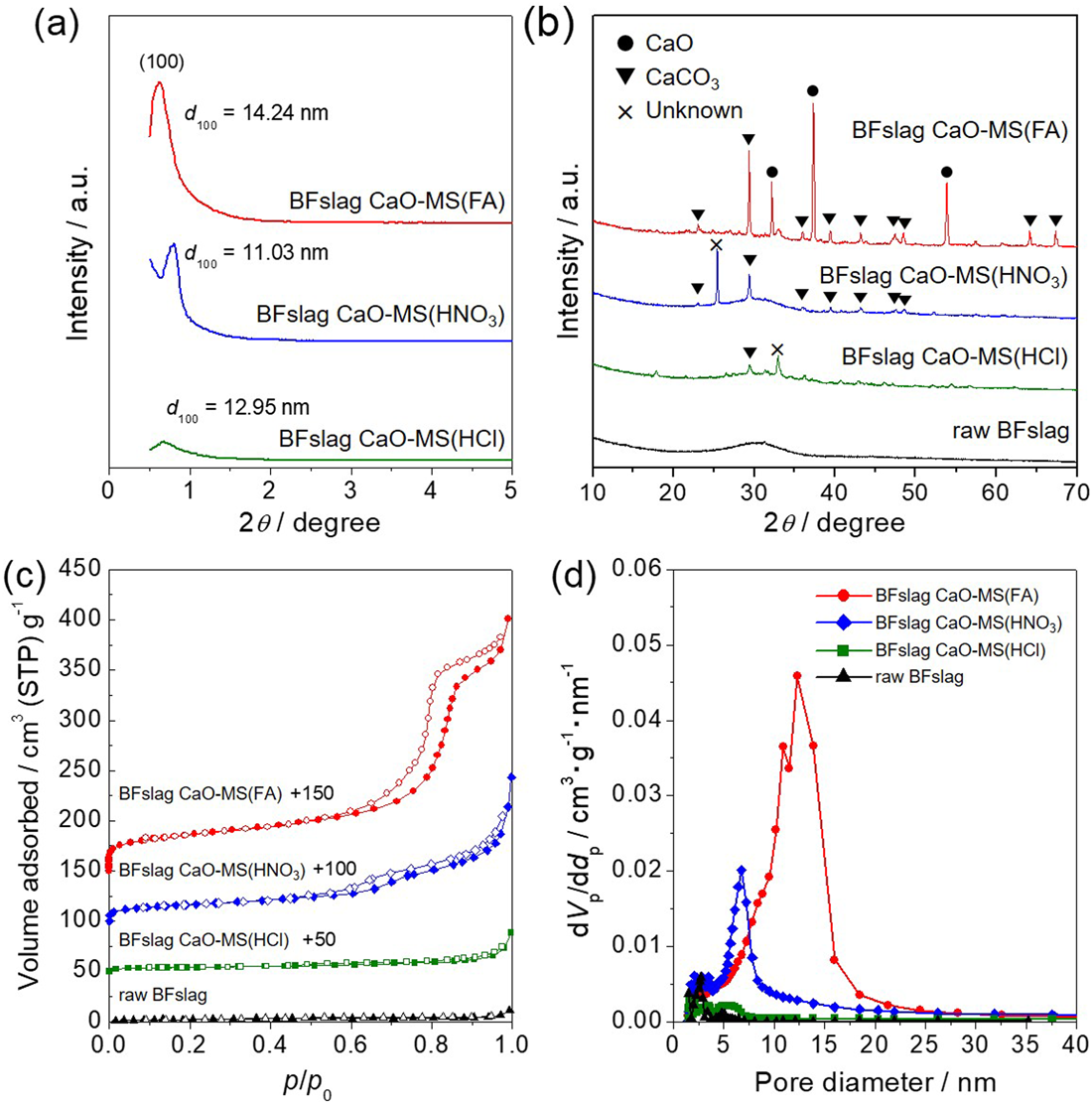
ギ酸を用いて合成したBFslag CaO-MS試料のXRDパターンには,CaOと大気中CO2との反応によって生成するCaCO3由来の回折ピークが観察された(Fig. 7(b))。低角度側のXRDパターンには,2θ=0.6–0.8°に規則配列したメソ細孔の(100)面に由来する特徴的なピークが見られたが,(110)および(200)面由来の回折ピークは観察されなかったことから無秩序配列したメソ細孔の形成が示唆された(Fig. 7(a))。一方,硝酸または塩酸を使用して合成した試料では,CaCO3相由来の回折パターンは確認されたものの,ギ酸を使用して合成した場合と比較して結晶性が低かった。また,ギ酸を用いて合成したBFslag CaO-MSはIV型のN2吸脱着等温線を示し,相対圧0.8付近での急激な立ち上がりが見られた(Fig. 7(c))。BET比表面積は128 m2/gと見積もられ,BJH細孔分布解析およびtプロット解析により,12.3 nmを平均直径とするメソ細孔と大きなメソ細孔容積(Vmeso=0.227 cm3/g)を持ち,発達したメソ細孔を有することが確認された(Fig. 7(d))。一方,硝酸を使用して合成した試料ではややメソ細孔の存在が確認されたものの,塩酸を使用して合成した試料ではメソポーラスシリカの形成は確認されなかった。これら酸による結晶構造,細孔構造の違いは,合成時の金属酸化物の沈殿プロセスが異なるためであると考えられる。酸解離定数の小さなギ酸を溶解剤として使用すると,溶出したCa2+イオンの多くはギ酸カルシウム(Ca(HCOO)2)を形成する。結果,水熱処理中にSi4+イオンとP123ミセル間の相互作用が可能になり,メソ細孔を持ったSiO2粒子が形成される。その後,ギ酸カルシウムが溶媒蒸発と大気焼成段階で結晶性CaOとして析出することでCaO –メソポーラスシリカ複合体が得られる。一方,酸解離定数の大きな硝酸および塩酸を溶解剤として使用すると,酸性水溶液中のCa2+イオンを捕捉することができず,これがメソポーラスシリカ粒子の成長を阻害するものと考えられる。これらの結果から,高炉スラグを高表面積なCaO –メソポーラスシリカ複合体に変換するには,ギ酸が溶解剤として最も適していることがわかった。
ギ酸を用いて合成したBFslag CaO-MSには,形状の異なる二種類の粒子が観察された。制限視野電子回折(SAED)およびEDXにより各粒子を分析したところ,粗大で無細孔の粒子からはCa,O,Cが検出されCaCO3であることが(Fig. 8(a)–(c)),多孔質状でアモルファス構造を有する粒子からは主にSi,Al,Oが検出されAlを含有したメソポーラスシリカであることが確認された(Fig. 8(d)–(f))。メソポーラスシリカ相には通常のSBA-15シリカに見られるような二次元ヘキサゴナル細孔構造は観察されず,無秩序に配列した小胞状のメソ細孔が観察された。これは母液中に存在するスラグ由来不純物金属の存在によって,細孔の規則配列が妨げられたためであると考えられる。また,化学分析よりBFslag CaO-MSの最終的な組成は,CaO(42.7 wt%), SiO2(36.9 wt%), Al2O3(15.2 wt%), MgO (3.11 wt%)であり,その他を不純物金属(Fe, Mn, Ti)が占めていた。これらは原料の高炉スラグの化学組成(CaO(40.1 wt%), SiO2(34.6 wt%), Al2O3(14.8 wt%), MgO (5.29 wt%))とほぼ同等である。金属原子数から計算した高炉スラグの正味のリサイクル効率は100%に近く,本研究で提案した変換プロセスにより高炉スラグに含まれる金属成分のすべてをリサイクルできたことを示している。
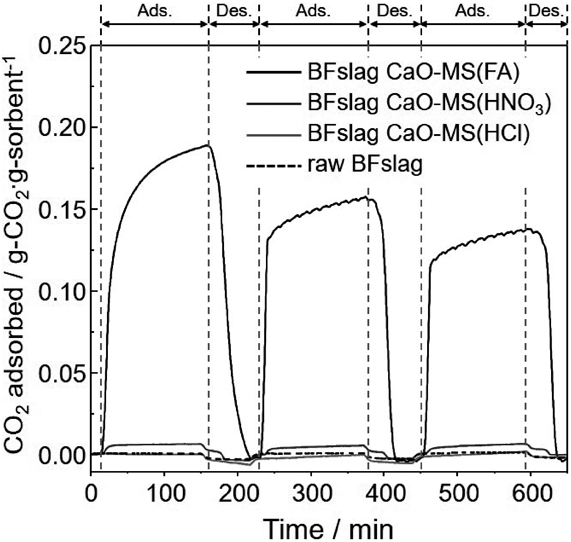
高炉スラグから合成したBFslag CaO-MSのCO2吸着性能は,排ガスを模擬した10%CO2/N2気流下で吸着実験を行い,熱重量分析装置を用いて重量変化を測定することにより評価した。Fig. 9に示すように,吸着温度700°Cにおいてギ酸を用いて合成したBFslag CaO-MSは吸着材重量あたり18.8 wt%のCO2吸着量を示し,750°C,100%N2に切り替えることで可逆的にCO2を脱離した。このCO2吸着量は試料に含まれるCaO単位重量あたりに変換すると46.9 wt%に相当し,市販のCaO (49.9 wt%)と同等のCO2吸着量である。硝酸を用いて合成した試料は微量のCO2を吸着したが(吸着材重量あたり0.7 wt%),塩酸を用いて合成した試料や原料の高炉スラグは同条件下でほとんどCO2吸着能を示さなかった。ギ酸を用いて合成したBFslag CaO-MSでのみ高いCO2吸着量と繰り返し吸着性能が得られた理由は,CO2との反応性を有する結晶性のCaO相が存在するためである。また,ギ酸を用いて合成したBFslag CaO-MS複合体は,CaOとメソポーラスシリカの物理的混合物(CaO+SBA-15)よりも高いCO2吸着量を示した。これは,メソポーラスシリカの細孔にCaO粒子が分散化することで露出CaO表面の割合が増加したためであると考えられる29,30)。さらに,細孔形成剤であるP123を使用せずに同様の手順で合成した類似体はBFslag CaO-MSと類似の化学組成と結晶相を有するにもかかわらず,CO2吸着量は半分程度であった。これは,メソポーラス構造の欠如,および表面積とメソ細孔容積の低さが原因であると考えられる。ナノメートルサイズの細孔が存在することで,CaO粒子が分散化するとともに粒子内のCO2ガスの拡散が促進され,吸着材としてのCO2吸着能力が向上したものと推察される。
4. 転炉スラグを原料としたCaO–Fe3O4–SiO2複合体の合成とCO2吸着への応用
高炉で製造した銑鉄は転炉に移され,生石灰などを入れ高圧の酸素を吹き込むことで銑鉄に含まれる不純物(C,P,S,Si,Mnなど)が取り除かれ,強靭な鋼が製造される。この際,鋼1トンに対し約110 kgの転炉スラグが副生する。日本の製鋼スラグ生産量のうち大部分をこの転炉スラグが占める(製鋼スラグ1342万トン中,転炉スラグ1072万トン(2019年度))1)。転炉スラグは,遊離したCaOを多く含むために高アルカリ性や膨張特性を有することや,FeO含有量が多い特異な組成を持つことから,高炉スラグに比べその利用用途が限られている。また,一部は埋立処分されており,転炉スラグの有効な再利用方法の開発は,鉄鋼産業の持続的な発展を目指す上で非常に重要な課題の一つである。
筆者らは,上述したギ酸を用いた溶解・水熱合成プロセスにより,転炉スラグをCO2吸着能を持つCaO系酸化物複合体へと変換することを試みた9)。転炉スラグを3.0 mol/Lのギ酸を用いて処理したところ,室温での攪拌でスラグ粒子の大部分は溶解したが,一部黒い粒子が酸に溶けずに残った。残存した粒子はFe3O4粒子であり,酸による溶解プロセスでは転炉スラグに含まれるFeO成分を溶出させることはできなかった。本手法では,これら未溶解のFe3O4粒子は,濾過等などのプロセスで除去せずに続けて合成を行った。得られた溶液にP123を加え,溶液に含まれるモル組成比がSi(転炉スラグに含まれるSi) : P123 : H2O=1 : 0.017 : 670となるように調整した。この溶液を,40°Cで24 h攪拌した後テフロンボトルに移し,100°Cで24 h保持して,溶解したSi4+種を縮合させ,シリカ骨格を形成させた。この懸濁液からエバポレーションにより水分を除去し,乾燥させた粉末を大気中600°Cで焼成することで,溶解していたCa2+イオンをCaO結晶として析出させた。このようにして3.0 gの転炉スラグから約3.4 gのCaO系酸化物複合体(Cslag CFS)を合成した。
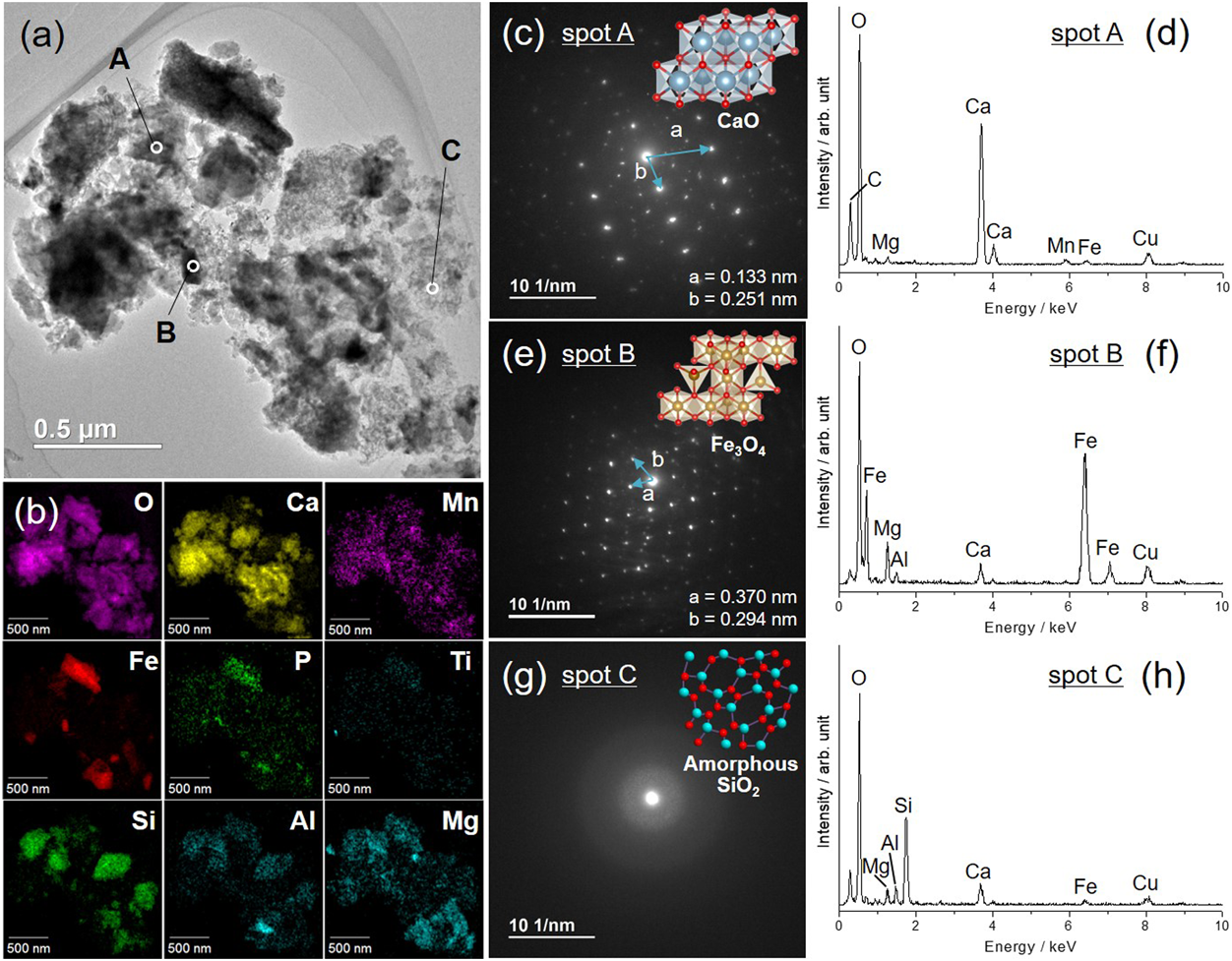
原料の転炉スラグからはFe3O4相とカルシウムフェライト(CaFe2O4)相が検出された。ギ酸を用いて転炉スラグから合成したCslag CFSにはCaCO3相とFe3O4相に帰属される回折パターンが見られた。このうち,Fe3O4相はギ酸での溶解時に溶け残った粒子である。また,Cslag CFSのXRDパターンにはBFslag CaO-MSと同様,2θ=0.6°付近に規則的なメソ細孔配列に帰属されるピークが見られた。一方,ギ酸の代わりに硝酸を用いた場合にはCaCO3相は形成されずカルシウムシリケート(Ca3MgSi2O8)相とFe3O4相が,塩酸を用いた場合には塩化カルシウム(CaCl2·2H2O)相とFe2O3相が確認された。高炉スラグを原料とした場合と同様に,CO2吸着能を有する結晶性CaO形成のためには,溶解酸としてギ酸を用いる必要があることがわかった。Cslag CFSのSTEM像ではコントラストや形状の異なる三種類の粒子が観察され(Fig. 10(a)),各粒子のSAED像およびEDXスペクトルから,大部分を占める固体粒子(スポットA)は結晶性CaO(空間群:Fm3−m)(Fig. 10(c), (d)),暗いコントラストの角張った粒子(スポットB)はFe3O4(空間群:Fd3−m)(Fig. 10(e), (f)),コントラストの低い微粒子凝集体(スポットC)はアモルファス状のSiO2(Fig. 10(g), (h))であると同定された。化学分析により求めたCslag CFSの最終組成と原料の転炉スラグの化学組成はほぼ一致しており,転炉スラグに含まれるほぼすべての金属成分が再利用されたことを意味している。元素マッピングの結果をもとにすると,微量不純物元素のうち,MnはCaO相に,P,TiはFe3O4相に,Al,MgはSiO2相にそれぞれ取り込まれやすいものと思われる(Fig. 10(b))。N2吸脱着測定において,転炉スラグからギ酸を用いて合成したCslag CFSはIV型の吸着等温線を示し,高炉スラグを原料とした場合と同様にメソ細孔を有していることが確認された。しかしながら,転炉スラグを原料とした試料では比表面積(SBET=64 m2/g),メソ細孔容積(Vmeso=0.066 cm3/g)ともに,高炉スラグを原料とした試料(SBET=128 m2/g, Vmeso=0.227 cm3/g)よりも低い値であった。これは転炉スラグのSiO2含有量(11.0 wt%)が高炉スラグのSiO2含有量(34.6 wt%)よりも少なく,試料重量当たりに含まれるSiO2粒子の割合が少なかったためである。
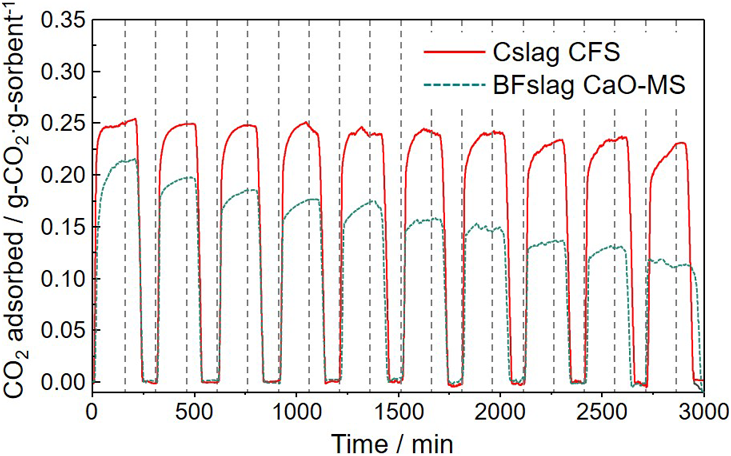
吸着温度700°C,10%CO2/N2流通下でCO2吸着性能を評価したところ,転炉スラグから合成したCslag CFSのCO2吸着量(23.4 wt%)は高炉スラグから合成したBFslag CaO-MSの吸着量(21.5 wt%)と比較して,初期吸着量で約1.1倍であった(Fig. 11)。これは転炉スラグのCaO含有量(43.8 wt%)が高炉スラグのCaO含有量(40.1 wt%)よりも多く,吸着材重量当たりに含まれるCaO粒子の割合が多かったためであると考えられる。原料の転炉スラグもカルシウムフェライト相の存在により約3.3 wt%のCO2吸着量を示したが,そのCO2吸着量はCslag CFSと比較するとはるかに低いものであった。また,P123を添加せずに合成した試料はCslag CFS と同じ化学組成・結晶構造を有するにもかかわらず,若干低いCO2吸着量を示した。P123を添加した場合には12 nm程度のメソ細孔の形成と表面積の増大が確認された。そのため,CaO粒子が高表面積メソポーラスシリカ担体上に高分散担持されたことや,粒子内のCO2ガスの拡散性が増したことで吸着量が向上したものと推測される。さらに,10サイクルの連続CO2吸脱着試験により吸着材の耐久性を評価したところ,BFslag CaO-MSでは10サイクル後のCO2吸着量が初期CO2吸着量の約55%まで低下したのに対し,Cslag CFSでは91%を維持した(Fig. 11)。XRD測定の結果,BFslag CaO-MSはメソポーラスシリカ相の含有量が多い(約37 wt%)ため,高温での繰り返し使用時に緻密なカルシウムシリケート相の形成が起こり,これに伴ってCO2吸着量が減少したことがわかった。一方,Cslag CFSではメソポーラスシリカ相の含有量が少ない(約12 wt%)ことに加え,CaOと反応性の低いFe3O4相が担体としての役割を担うことでCaO相のカルシウムシリケート相への相転移やCaOどうしのシンタリングが抑制され32),再利用性が向上したことがわかった。
上記のように,複雑な化学組成を有する高炉スラグや転炉スラグを原料としても,CO2吸着能を有する機能性材料が合成可能であることを実証した。とりわけ,転炉スラグを用いて合成した吸着材は高炉スラグから合成した吸着材よりも高いCO2吸着量および高温での再生能力を示した。開発したスラグ変換プロセスは簡便かつ経済的であることから,本プロセスで合成した吸着材は,鉄鋼産業や石炭火力発電所などから排出される排ガスからのCO2吸着分離にも適用可能であると考えられる。実用化に向けては,今後,実際の排ガスを模擬した条件での吸着実験や合成に係る経済性評価が必要であるが,本プロセスは現在鉄鋼産業が直面している『廃スラグの有効利用』と『CO2排出量の削減』という二つの重要な問題に貢献しうる技術として今後のさらなる展開が期待できる。
鉄鋼スラグを出発原料とした資源循環プロセスの構築を目的として,これまで筆者が取り組んできた研究内容を紹介した。鉄鋼スラグは複雑な化学組成を有しているが,各含有成分の化学的特性を利用することで,より付加価値の高い材料へと変換することができる。本研究では,適切な溶解酸を利用することでCa2+イオンを捕捉・分離し,その後,ゼオライトやメソポーラスシリカの骨格形成を促す逐次的な結晶化プロセスにより機能性酸化物複合体へと変換することに成功した。スラグ変換プロセスの要は,複雑に絡み合った金属–酸素結合を切断し,いかに効率的に目的とする化合物に組み替える(再構成する)かである。そこでは,各含有元素の持つ酸や水酸化物イオンとの反応性,各元素どうしの反応性・親和性をいかに制御するかが鍵となる。一連の研究はいずれも,国内で大量に副生成する鉄鋼スラグの有効利用のみならず,高付加価値材料の安価な製造方法の確立も目的としている。また,このような物質変換プロセスは,鉄鋼スラグのみにとどまらず他の無機産業副産物へも応用可能であると考えられる4,5,33)。これらの技術が実用化に至るには,今後,現行の製造コストを下回る低コスト化,合成プロセスの簡略化,廃棄物利用による弊害リスク(不純物混入による製品性能の低下や化学組成の不均質性の問題)のさらなる低減が必要である。
また,本研究で得られた酸化物複合体は,含有酸化物の化学特性に基づく吸着能を示し,環境保全用の吸着材やCO2吸着材として利用可能である。その吸着性能は試薬から合成した純正品とほぼ同等の吸着能を示すが,スラグ由来不純物元素を含むため選択性や純度が要求される場面での利用には適さない。このため,“質”よりも“量”が必要とされる環境保全分野での利用がふさわしいと筆者らは考える。従来用いられている高価な吸着材の代替材料として鉄鋼スラグから合成した安価な吸着材が利用できれば,鉄鋼スラグを出発原料とする新しい資源循環プロセスの創成につながるものと期待している。
謝辞Acknowledgments
本稿に記した研究成果は科学研究費補助金(21K05147),新井科学技術振興財団平成30年度研究助成,高橋産業経済研究財団令和2年度研究助成,八洲環境技術振興財団 2020年度研究開発・調査助成,および日本鉄鋼協会令和3年度鉄鋼研究振興助成の支援の下なされたものであり,ここに記して感謝申し上げる。特に,鉄鋼スラグを原料に用いたCO2吸着材合成の研究に関しては,花木愛子氏(当時大阪大学大学院工学研究科博士前期課程)の努力によって成し得たものであり,記して感謝申し上げる。
引用文献References
1) The Japan Iron and Steel Federation, Nippon Slag Association, The Annual Report of Statistics of Iron and Steel slag 2019, http://www.slg.jp. (Retrieved on Feb. 24th, 2022)
2) B. Das, S. Prakash, P. S. R. Reddy, V. N. Misra, Resour., Conserv. Recycl., 50, 40(2007).
3) S. Kumar, R. Kumar, A. Bandopadhyay, Resour. Conserv. Recycl., 48, 301(2006).
4) M. Balakrishnan, V. S. Batra, J. S. J. Hargreaves, I. D. Pulford, Green Chem., 13, 16(2011).
5) J. A. Bennett, K. Wilson, A. F. Lee, J. Mater. Chem. A, 4, 3617(2016).
6) Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori, H. Yamashita, J. Mater. Chem., 19, 7263(2009).
7) Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori, H. Yamashita, Chem. Lett., 38, 626(2009).
8) Y. Kuwahara, A. Hanaki, H. Yamashita, Green Chem., 22, 3759(2020).
9) Y. Kuwahara, A. Hanaki, H. Yamashita, ACS Sustainable Chem. Eng., 10, 372(2022).
10) Y. Kuwahara, S. Tamagawa, T. Fujitani, H. Yamashita, J. Mater. Chem. A, 1, 7199(2013).
11) Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori, H. Yamashita, J. Mater. Chem., 20, 5052(2010).
12) Y. Kuwahara, S. Tamagawa, T. Fujitani, H. Yamashita, Bull. Chem. Soc. Jpn., 89, 472(2016).
13) Y. Kuwahara, H. Yamashita, ISIJ Inter., 57, 1657(2017).
14) Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori, H. Yamashita, Catal. Sci. Technol., 2, 1842(2012).
15) Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori, H. Yamashita, ChemSusChem, 5, 1523(2012).
16) Y. Kuwahara, H. Yamashita, J. CO2 Util., 1, 50(2013).
17) Y. Sugano, R. Sahara, T. Murakami, T. Narushima, T. Iguchi, C. Ouchi, ISIJ Inter., 45, 937(2005).
18) Y. Kuwahara, T. Ohmichi, K. Mori, I. Katayama, H. Yamashita, J. Mater. Sci., 43, 2407(2008).
19) V. K. Jha, M. Matsuda, M. Miyake, J. Ceram. Soc. Jpn., 116, 167(2008).
20) Y. Kondo, T. Goto, Y. Kuwahara, T. Sekino, H. Yamashita, manuscript in preparation.
21) Ministry of the Environment, Government of Japan, Japan’s National Greenhouse Gas Emissions, https://www.env.go.jp. (Retrieved on Feb. 24th, 2022)
22) C. Xu, D. Q. Cang, J. Iron Steel Res. Int., 17, 1(2010).
23) K. Tanaka, Energy Policy, 51, 578(2012).
24) B. Dutcher, M. Fan, A. G. Russell, ACS Appl. Mater. Interfaces, 7, 2137(2015).
25) M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox, N. Mac Dowell, Energy Environ. Sci., 11, 1062(2018).
26) S. Tian, J. Jiang, Z. Zhang, V. Manovic, Nat. Commun., 9, 4422(2018).
27) T. A. Adams II, L. Hoseinzade, P. B. Madabhushi, I. J. Okeke, Processes, 5, 44(2017).
28) D. P. Hanak, E. J. Anthony, V. Manovic, Energy Environ. Sci., 8, 2199(2015).
29) C. H. Huang, K. P. Chang, C. T. Yu, P. C. Chiang, C. F. Wang, Chem. Eng. J., 161, 129(2010).
30) F. Yan, J. Jiang, K. Li, S. Tian, Z. Liu, J. Shi, X. Chen, J. Fei, Y. Lu, ACS Sustainable Chem. Eng., 4, 7004(2016).
31) D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka, G. D. Stucky, Science, 279, 548(1998).
32) S. Tian, J. Jiang, D. Hosseini, A. M. Kierzkowska, Q. Imtiaz, M. Broda, C. R. Müller, ChemSusChem, 8, 3839(2015).
33) J. Wang, Y. Yang, Q. Jia, Y. Shi, Q. Guan, N. Yang, P. Ning, Q. Wang, ChemSusChem, 12, 2055(2019).