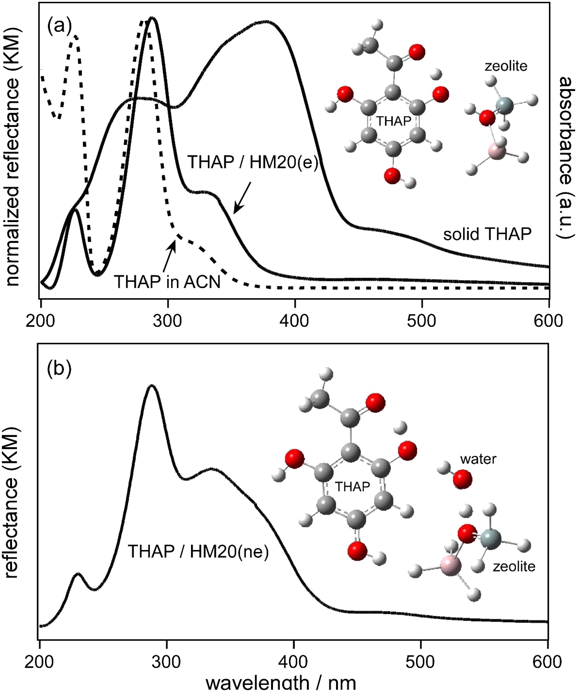
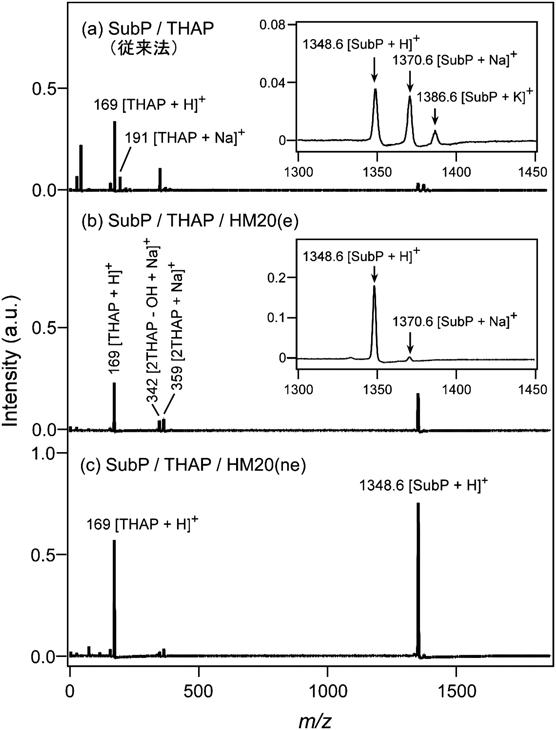
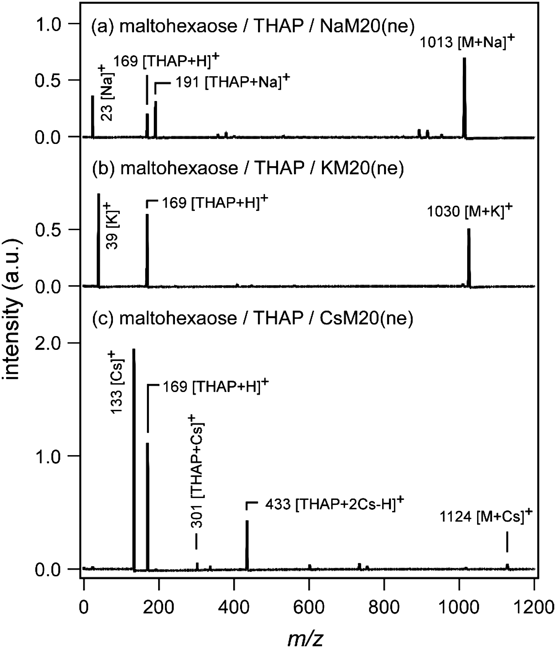
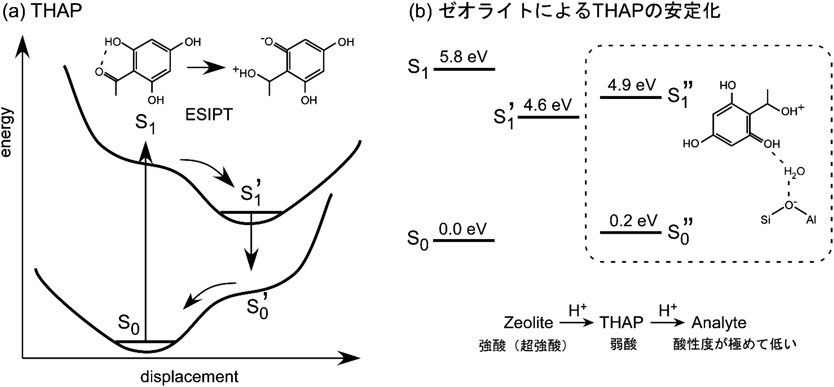
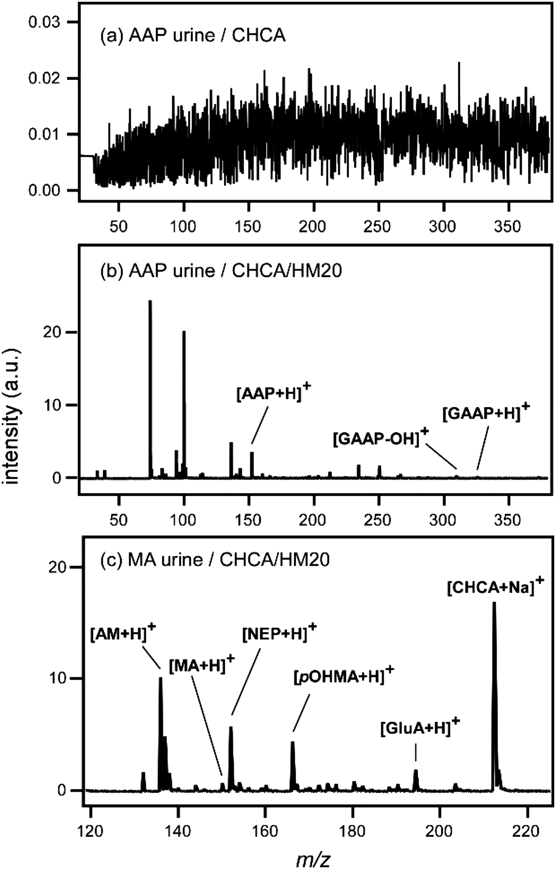
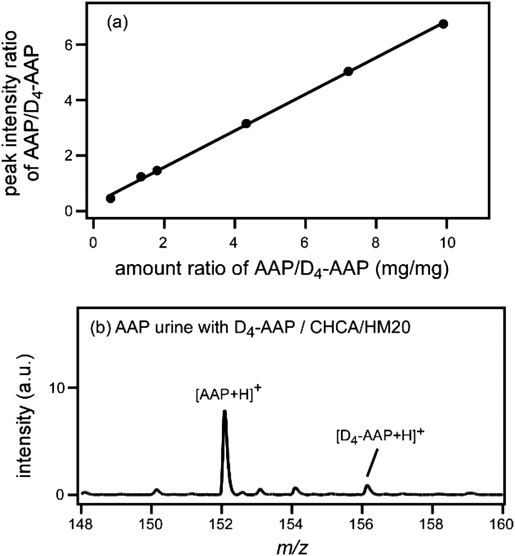
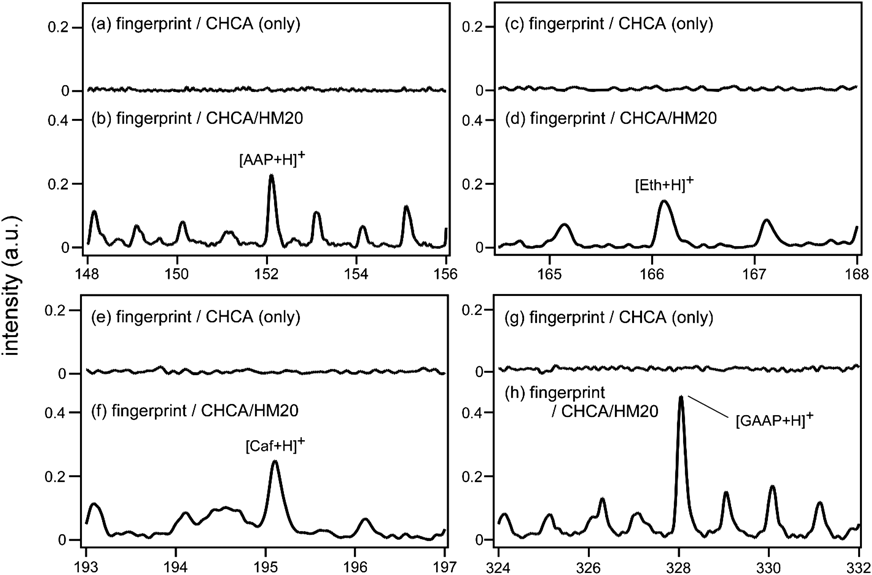
レーザー脱離イオン化質量分析法へのゼオライトの利用Application of Zeolite to Laser Desorption Ionization Mass Spectrometry
1 東洋大学大学院理工学研究科応用化学専攻Department of Applied Chemistry, Graduate School of Science and Engineering, Toyo University ◇ 〒350–8585 埼玉県川越市鯨井2100
2 東洋大学バイオナノエレクトロニクス研究センターBio-Nano Electronics Research Centre, Toyo University ◇ 〒350–8585 埼玉県川越市鯨井2100
3 警視庁科学捜査研究所Criminal Investigation Laboratory, Metropolitan Police Department ◇ 〒100–8929 東京都千代田区霞が関2丁目1番1号
4 放送大学教養学部Materials, Energy and Environmental Sciences, The Open University of Japan ◇ 〒261–8586 千葉県美浜区若葉2–11
受理日:2021年10月20日Accepted: October 20, 2021
発行日:2022年1月31日Published: January 31, 2022