2020年,日本政府は経済・環境の好循環を国の成長戦略の柱として,2050年までに温室効果ガスの排出を全体としてゼロにする「2050年カーボンニュートラル」に挑戦することを宣言した。炭素循環を実現しながらエネルギーを得るためには,地球系外からエネルギーを得ることが不可欠であり,太陽光エネルギーの利用はまさに中核を成す課題である。太陽光エネルギーを電気エネルギーに変換する太陽電池は,1954年のSi太陽電池の発明以降急激に普及し,現在ではペロブスカイト系などのいくつかのシリーズの太陽電池が,高いエネルギー変換効率でしのぎを削っている1)。しかし,人口密集地へのエネルギー供給を行うためには,太陽光エネルギーを貯蔵・輸送する必要があり,この場面においては従来の化石資源のような化学エネルギーに変換する,いわゆる「人工光合成システム」を開発する必要がある2,3)。1972年の本田–藤嶋効果の発見4)から半世紀がたち,現在ようやく実用レベルの効率(太陽光–水素エネルギー変換効率~10%)を発揮する「人工光合成システム」の開発ができてきた5–7)。
「人工光合成システム」の心臓部は光触媒である。長期安定性の面から,太陽電池にも用いられる無機半導体材料(TiO2,WO3,Ta3N5など)が光触媒材料として頻繁に用いられる。半導体がバンドギャップエネルギー以上の光子エネルギーを吸収して,励起電子・正孔を生成するところまでは太陽電池と同様の動作原理である。太陽電池と本質的に異なる点は,光触媒には水の分解反応を触媒する助触媒が担持されており,励起電子・正孔が助触媒に到達することで化学反応が進行する点である8)。化学反応は多電子過程であることから,励起電子・正孔の逆電子移動(再結合)によるエネルギー損失が起こりやすく,これが低い効率の原因となる。近年,堂免らのグループはAlドープSrTiO3光触媒を用いた水の全分解反応について,特定結晶面への助触媒担持による外部量子効率96%での水の全分解反応を報告した9)。SrTiO3内部での励起電子・正孔の移動に異方性があることによって,励起電子・正孔の逆電子移動の寄与をほぼ0%にできることを見出した。しかしながら,SrTiO3は太陽光エネルギーの主成分である可視光を吸収しない光触媒材料であるため,以下の戦略が必要となる。
- (a)励起電子・正孔の異方性移動が起こる可視光駆動型光触媒を開発する。
- (b)太陽光中の可視光を紫外光に変換する材料を開発し,従来の紫外光応答光触媒を駆動させる。
本稿の前半部分(第2,3章)では,層状ナノ材料の精密設計と層間ナノ空間への発光性有機分子の精密配置を利用した,これらの戦略に資する材料の開発について述べる。具体的に第2章では,チオールエンクリック反応によってチタン酸ナノシート光触媒とタングステン酸ナノシート光触媒の交互積層構造を創出し,層間および外表面にRu(bpy)32+とPt(terpy)錯体を配置した材料について述べる。この材料を用いた水素生成光触媒反応に成功し,可視光照射によるRu(bpy)32+を介したタングステン酸からチタン酸へのベクトル電子移動をナノ秒分解のダイナミクス解析によって実証した。第3章では,疎水化した粘土層間に有機分子を充填することによる,従来困難であった水・酸素共存中で緑色光から青色光への短波長変換が可能なフォトン・アップコンバージョン材料を開発した事例について述べる。この材料を用い,太陽光中の主成分である長波長光を利用して,短波長光下でのみ作用する光触媒を駆動させることに成功した。
地球に降り注ぐ太陽光を使ったエネルギー製造に対して,電力から作り出した光を巧みに利用した情報伝達・知覚空間の創出・センシング技術も,光化学研究の重要なキーテーマである。我が国は,2016年に発表した第5期科学技術基本計画において,目指すフィジカル空間とサイバー空間が高度に融合したシステムによって経済発展と社会的課題解決を両立する人間中心の社会(Society 5.0)を提言した。この社会構想において,人間の視覚を利用して空間相互での情報交換を可能とするディスプレイデバイスや,遠隔的に情報を近くする光センサーの重要性がより一層高まる。例えば,有機物の電界発光(EL)現象を利用した有機ELディスプレイは,高画質(高精細・高コントラスト比),高速応答性,フレキシブル(曲げられる)性を有し,従来のディスプレイを遥かに凌ぐ情報交換機能を次世代スマートフォン・テレビに与えることができる10)。また,生体親和性の高い発光物質は,高度な医学検査を可能とするセンサーとして機能する11)。発光波長・色純度は,この緩和に伴う分子形状や電子状態の変化などによって決まる。これらの発光挙動制御に対するこれまでの戦略は,「特別な有機分子を設計・合成する」ことに大きく依存してきた。長年の研究蓄積によって膨大な数の発光体分子が開発され,発光挙動制御のための分子設計に関する学理も十分に整っている12)。しかしながら,有機発光材料開発は次の二つの根本的な問題によって頭打ちになりつつある。
- 無機材料と比較して色純度が低いため,色再現性が低い。
- 複雑な有機分子の生産には高環境負荷の化学プロセスを必要とすることが多い。
以上の課題を克服する,有機EL材料開発のための新たなパラダイム・戦略が切望されている。
一方,ここ30年間余りで,結晶性ケイ酸塩が作り出すナノ空間に光活性な分子を導入し,触媒活性や発光挙動を制御する研究が数多く行われてきた13,14)。非常に先駆的な例として,和田らはNa-FAUゼオライト細孔内にFe錯体やMo錯体などの光活性分子を導入し,オレフィン転換反応に対する光触媒活性を実証した15,16)。更に2000年代以降には,Ship-in-a-bottle法などを用いて導入された様々な金属錯体の特異な発光特性や,ゼオライト細孔内でのみ実現されるAgクラスターによる発光挙動が多く報告された17)。
他方,有機発光体のゼオライト細孔内導入については,2000年前後に橋本ら18),Cormaら19),Brémardら20)がそれぞれ,Na-FAU,Na-MOR,Na-MFIゼオライトへの溶液吸着法による青色発光分子:アントラセン導入を報告している。ゼオライトの閉じ込め効果やAlサイトとの電子交換による特異な電子状態のアントラセン安定化や,狭いケージ内での二分子発光過程:エキシマー発光による緑色発光が観測されている。これらの事実は,ゼオライト細孔内にアレーン誘導体を導入することで,発光状態をドラスティックに変化できることを示唆している。しかし,興味深い発光特性を示す有機分子はアントラセンのように中性・疎水性のアレーン誘導体であるため,狭い細孔に大量導入することは困難である。そのため,マクロに興味深いゼオライト内での有機分子の発光挙動(レーザー発振21,22),偏光発光23)など)が観測される系は,筆者が知る限りでは,カチオン性有機分子を使った例のみである。
本稿の後半(第4章)では,粘土層間やゼオライトなどのナノ空間に対する無置換アレーン誘導体の新たな導入法として,カチオン交換によって導入したアレーンアンモニウムを粘土・ゼオライトの触媒作用によって細孔内で脱アミン化し,狭い細孔内に大量の無置換アレーン誘導体を生じる方法について述べる。この「細孔内脱アミン化法」を利用すると,粘土層間には熱・光安定なアリールラジカルカチオンが高密度に充填された。このラジカルカチオンを開始剤としたスチレンの重合反応によって高分子末端に直接アリール基が結合した,光機能性ポリスチレンを得ることに成功した。また,「細孔内脱アミン化法」を利用してMOR,FAUゼオライト細孔内に無置換アントラセンを導入し,それぞれ高純度青色発光体および白色発光体を創出することに成功した。
2. 光触媒ナノシート積層型ベクトル電子移動系の構築
太陽光中の大部分を占める可視光を利用した人工光合成系構築に向けて,低いエネルギーの光子で駆動する二種類の半導体光触媒を利用したZ-Scheme型の電子移動系が研究されている(Fig. 1(a))24)。これら二種類の半導体光触媒粒子を水に分散,あるいは基板に固定し,粒子間で起こる光誘起電子移動を駆動力とした水分解反応を行う。しかし,光触媒同士のランダムな構造に起因して,望まない逆方向の電子移動も同時に起こり効率を低下させることが知られている。
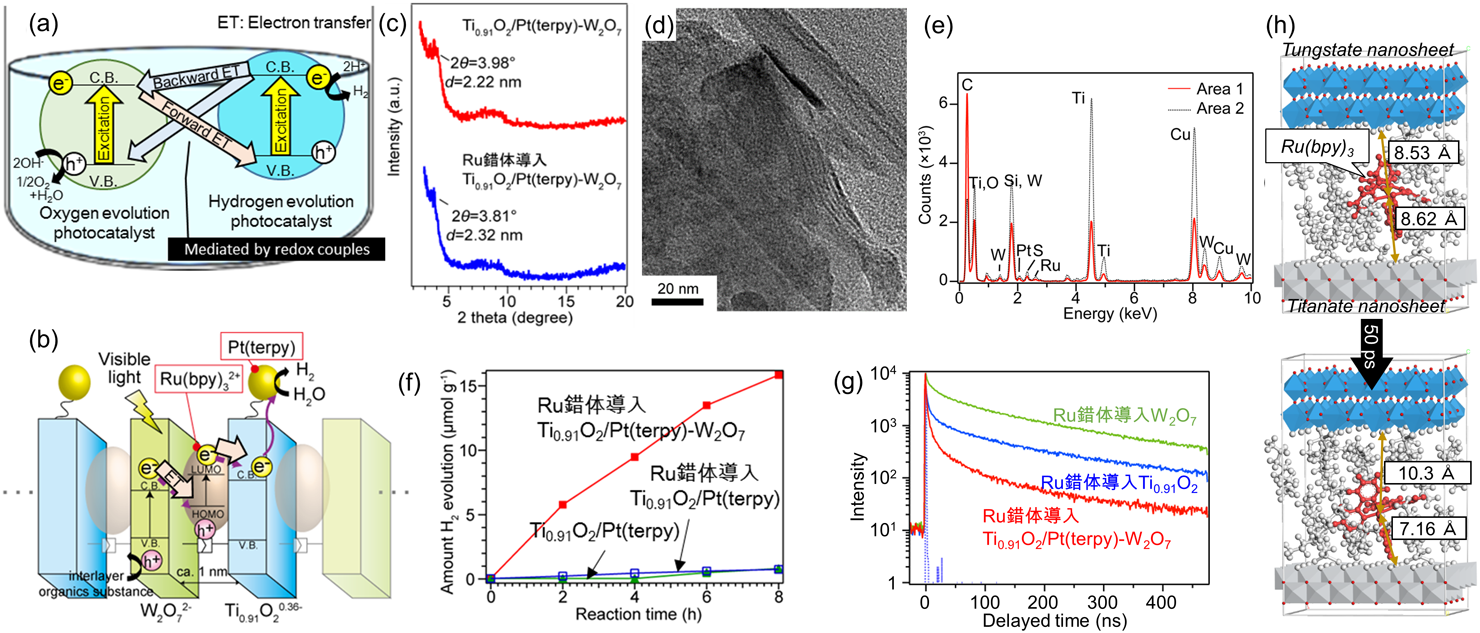
本研究では,逆電子移動が抑制されたベクトル的な光誘起電子移動系の構築を目指した無機ナノ構造設計を行った。チタン酸ナノシート(Ti0.91O2)とタングステン酸ナノシート(W2O7)にそれぞれ修飾したチオール基とアルケニル基のチオール–エンクリック反応によって交互積層構造25–27)を創出し,金属錯体(Ru(bpy)32+, Pt(terpy))を場所選択的に固定化した(Fig. 1(b))28)。理想とする順方向の電子移動パスは,可視光照射によって励起状態となるRu(bpy)32+のLUMOレベルがチタン酸の伝導帯に電子を供与し,Ru(bpy)32+の空のHOMOレベルにタングステン酸の伝導帯電子が移動することである。結果として,より高い電位を持つチタン酸の伝導帯に電子を蓄積することができ,チタン酸ナノシートの縁辺にあらかじめ固定化したPt(terpy)分子上で水を還元することができる。
2.1 Ru(bpy)32+,Pt(terpy)修飾チタン酸/タングステン酸ナノシート交互積層構造による光触媒作用
層状チタン酸を出発材料とし,各層の縁辺へのPt(terpy)の固定化と,層間面へのチオール基の修飾を施したのち,DMF中でナノシートに剥離した。このチタン酸ナノシートを,層状タングステン酸の剥離によって得られたアルケニル修飾タングステン酸ナノシートとのチオール–エンクリック反応によって交互積層構造を得た。層間に対するRu(bpy)32+の導入はイオン交換法によって行った。材料のキャラクタリゼーションは主にX線回折と電子顕微鏡観察によって行った。Ti0.91O2/Pt(terpy)-W2O7の2θ=3.98°に発現した周期的なナノシートの積層構造由来のX線回折ピークは,Ru錯体導入後に2θ=3.81°にシフトした(Fig. 1(c))。つまり,Ru錯体が層間に導入されたことで層間距離が押し広げられたと考えられる。Ru錯体導入Ti0.91O2/Pt(terpy)-W2O7のTEM像から,層状構造由来の縞模様が観察された(Fig. 1(d))。この視野におけるEDXスペクトルには,Ti,Pt,W,Ruのピークが発現したため,この縞模様は交互積層構造に由来するものであると帰属できる(Fig. 1(e))。得られた交互積層構造の粉末を純水に分散し,可視光照射(λ>420 nm)下での水素発生を実証した(Fig. 1(f))。Ru錯体を導入した交互積層体は,Ru錯体を導入したPt錯体修飾層状チタン酸(Ti0.91O2/Pt(terpy))と比較して高い水素発生光触媒能を示した。
交互積層構造中で起こるベクトル電子移動を立証するためには,各電子移動ステップを定量的に評価する必要がある。まず,光励起状態のRu(bpy)32+のLUMOレベルからの電子移動ダイナミクスを蛍光減衰から求めた(Fig. 1(g))。R(bpy)32+のLUMOレベルからチタン酸伝導帯への順方向の電子移動の速度は1.16×108 s−1と見積もられ,タングステン酸伝導帯への逆電子移動の速度1.02×107 s−1よりも10倍以上速いことが分かった。その理由が,層間でRu(bpy)32+がチタン酸ナノシート側に局在化して存在するためであることを,分子動力学法による層間での分子種のふるまいをシミュレーションすることで明らかにした(Fig. 1(h))。タングステン酸伝導帯から光励起状態のRu(bpy)32+の空のHOMOレベルへの電子移動については,タングステン酸伝導帯に蓄積された電子量を近赤外拡散反射法によって見積もる方法で実証することができた27)。
2.2 Eu3+ドーピングによるナノシート間での逆電子移動ダイナミクス計測
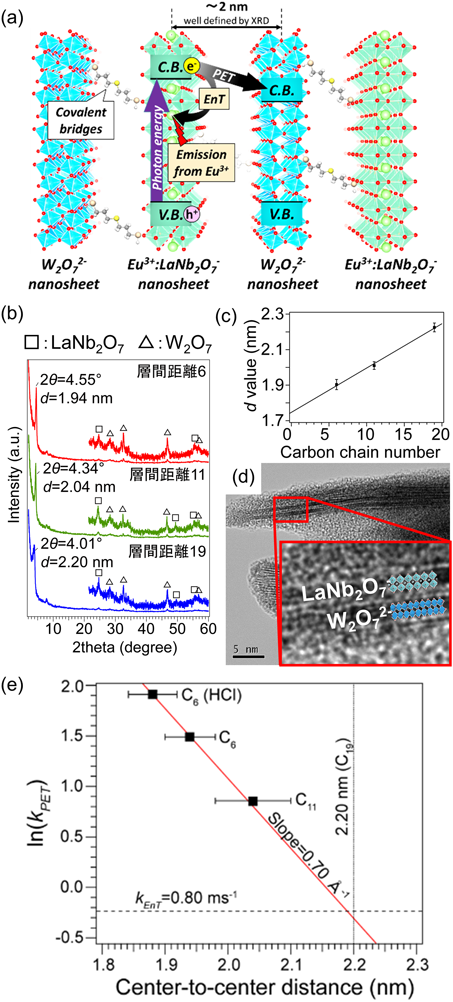
半導体光触媒間での逆電子移動は,Z-Scheme型電子移動系の最大のボトルネックである。チタン酸/タングステン酸ナノシート交互積層体では,チタン酸からタングステン酸への電子移動過程がこの逆電子移動に相当する29)。このダイナミクスを速度論的に計測することを目的として,タングステン酸ナノシートを発光性Eu3+をドープしたLaNb2O7と交互積層し,発光減衰から半導体間光誘起電子移動の速度を厳密に決定した(Fig. 2(a))30)。LaNb2O7を光励起させて生じる伝導帯電子は,エネルギー差によってタングステン酸の伝導帯に移動する。ナノシートにチオール基とアルケニル基を固定する際に使用した分子の炭素鎖長を変化させることで,隣り合うナノシート間距離をオングストローム単位で系統的に変化させ,ナノシート間で起こる電子移動速度の変化を調べた。これによってナノシート間電子移動の機構を明らかにし,逆電子移動ダイナミクスを概算することとした。
ナノシートに固定したチオール基とアルケニル基の炭素鎖長を6,11,19と変化させると,交互積層構造の隣り合うナノシートを回折面とするXRDピークは2θ=4.55°,4.34°,4.01°と段階的に変化した(Fig. 2(b))。そのd値は炭素鎖長に従って直線的に増大し,ナノシート間距離(隣り合うナノシート間の中心間距離と定義)を精密に制御することに成功した(Fig. 2(c))。TEM像には交互積層構造を示す明確な縞模様が観測された(Fig. 2(d))。Eu3+のミリ秒発光減衰測定の結果から,ナノシート間での電子移動の速度を見積もった。Fig. 2(e)に,電子移動速度をナノシート間距離に対してプロットした。プロットは良い直線関係を示し,傾きが0.70 Å−1であることから,この電子移動機構がスルースペース型トンネリング機構であると結論した。また,ナノシート間距離が十分に大きい(2.2 nm)とき,電子移動反応は起こらなくなった。これは,LaNb2O7からタングステン酸への電子移動の速度が,ドープされたEu3+へのエネルギー移動の速度よりも遅くなったため,ナノシート間電子移動が起こらなくなったためと考えられる。
以上の結果から,電子供与側のナノシートにドーピングしたEu3+の発光減衰に基づき,ナノシート間で起こる動的な電子移動反応のメカニズムがスルースペース機構であると結論した。この結果を単純にチタン酸/タングステン酸ナノシートの系に持ち込んで議論することはできないが,十分なナノシート間距離を生み出すことで,逆電子移動過程を抑制することができる。望月ら31)は,本研究と同様の手法で合成したチタン酸/タングステン酸交互積層体において,ナノシート間電子移動によってタングステン酸伝導帯に蓄積された電子数(eET/g)をAg還元反応による滴定によって求め,次の関係式が成り立つことを示した。
ここで,d′はナノシート間距離からナノシートの厚みを減算した層間距離を表す。今回合成したチタン酸/タングステン酸交互積層体のナノシート間距離は1.46 nmであり,タングステン酸伝導帯への電子蓄積の速度は2.7×1017 electrons min−1 gsample−1である。この電子量は,TiO2粒子/WO3粒子物理混合品に紫外光を照射したときに蓄積される電子量(WO3の自己励起によって蓄積される電子量に相当)と同等であることから,チタン酸伝導帯からタングステン酸伝導帯への電子移動はほとんど起こらないと結論することができる。
第2章では,数Å程度の距離で配置したナノシート積層構造に対するナノ空間への金属錯体修飾によって,逆電子移動を抑制したベクトル電子移動系の創出について述べた。特に本系は,ランダムな配置を持つ従来のZ-Scheme型電子移動系と異なり,光誘起電子移動の速度論を素反応過程ごとに取り出した議論を実現した。しかしながら,逆電子移動を完全に抑制することはできず,これらは光触媒能を低下させる要因となる。水素生成の結果から見積もられる,衝突したλ=420~600 nmの光子数当たりの量子効率は0.1%にも満たない。光触媒の複合構造を用いる限りは逆電子移動を完全に抑制することは難しく量子効率を低減してしまう。根本的な解決のためには単一の光触媒を利用した人工光合成システムが肝要であり,第3章に繋ぐ。
人工光合成系がZ-scheme電子移動系を利用する理由は,低い光子エネルギーを持つ長波長光(>500 nm)を有効利用するためである。しかし前述の通り,これらの光触媒の量子効率はそれほど高くない。SrTiO3に代表される高量子効率を示す光触媒は,長波長光では駆動せず大部分の太陽光を利用できないジレンマがある。そこで想起される「人工光合成システム」開発のもう一つの戦略は,太陽光中の可視光を紫外光に変換する材料を開発し,従来の水分解反応に高活性な光触媒をそのまま用いることである。
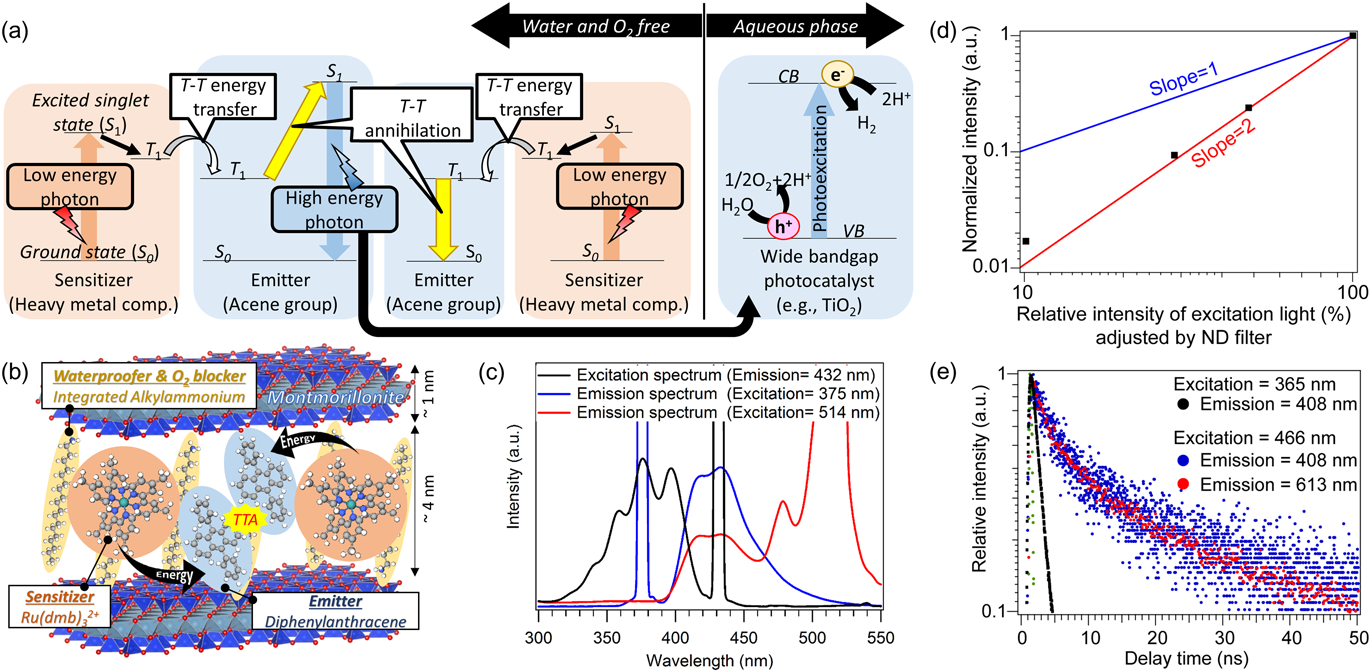
太陽光中の可視光を紫外光に変換するための手段として,特に近年,有機色素の三重項–三重項消滅(TTA)過程に基づくフォトン・アップコンバージョン(TTA-UC)が注目されている。TTA-UCでは,①長波長光吸収によって生成した三重項励起(T1)状態の増感剤2分子から,②エネルギー移動によってT1状態の受容体2分子が生成し,③これらの分子のTTAによって生じる一重項励起(S1)状態の受容体分子から短波長発光が起こり,光触媒に利用することができる(Fig. 3(a))。しかし,T1状態の分子は酸素によって失活しやすく,水分解を目的とした人工光合成系への導入は困難である。本研究では,アルキルアンモニウムカチオンによって疎水化された層状化合物の層間に,増感剤としてルテニウムトリスジメチルビピリジン(Ru(dmb)32+),受容体としてジフェニルアントラセン(DPA)を導入し,水および酸素による消光を防いだTTA-UC発光粒子(UCP)の開発を目指した(Fig. 3(b))32)。
3.1 天然モンモリロナイト層間でのフォトン・アップコンバージョンの実証
クニミネ工業(株)から購入した天然モンモリロナイトをジオクタデシルジメチルアンモニウム(DODMA)と反応させ,層間拡張・疎水化した。この試料をRu(dmb)32+のアセトニトリル溶液に24 h分散した。得られた試料をDPAのトルエン溶液に分散した。得られたUCPの励起発光スペクトルをFig. 3(c)に示す。432 nm発光測定時の励起スペクトルおよび375 nm励起光下の発光スペクトルは,DPAの自己励起発光に対応するスペクトルである。Ru(dmb)32+を励起する514 nm光照射下での発光スペクトルにおいて,375 nm励起光下で観測されたDPA由来の発光ピーク(419 nmと432 nm)と同様のスペクトルが観測された。この結果は,514 nm光が419~432 nm光に変換されたこと示す。励起光強度に対する発光強度依存性(Fig. 3(d))を測定したところ,432 nmにおける発光強度は514 nm励起光の強度に対して2次の関係を示した。つまり,2光子吸収による分子励起によって1光子が生成したことを示す。更に,466 nmパルスレーザー励起による408 nm発光減衰速度は,Ru(dmb)32+のT1→S0緩和由来の613 nm発光の減衰と良い一致を示した(Fig. 3(e))。すなわち,466 nm励起光照射下における408 nm発光過程は,Ru(dmb)32+のT1状態からのエネルギー移動によって引き起こされるものであることを示す。以上の結果から,光励起によって生じるRu(dmb)32+のT1状態からのエネルギー移動によってDPAのT1状態が生じ,2分子のT1状態のDPA間での三重項–三重項消滅過程を経て生じたS1状態のDPAが緩和する過程で432 nm光を発する,TTA-UCが起こったと結論できる。
3.2 粘土層間フォトン・アップコンバージョンによる水懸濁系の光触媒反応の駆動
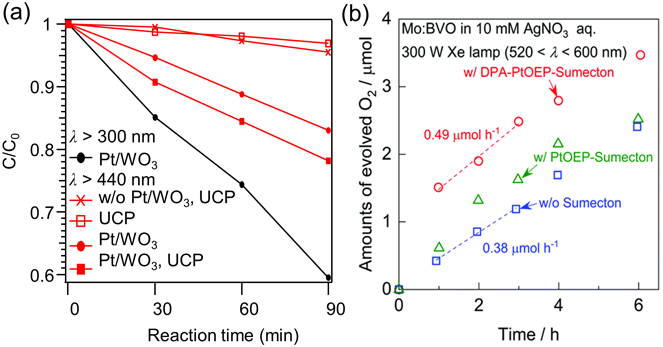
得られたUCPを光触媒とともに水に分散し,TTA-UCによって生じる短波長光を利用して光触媒を駆動させる実証実験を行った。まず,UCPをPt/WO3光触媒とともにローダミンB水溶液(1 mM)に分散し,Xeランプを照射してローダミンB光分解反応への実証実験を行った。ローダミンBの光分解実験では,ローダミンBの初期濃度(C0)からの変化率(C/C0)をUV-Vis吸収スペクトルによって決定し,Fig. 4(a)にプロットした。Pt/WO3単体はλ>300 nm光照射下で自己励起によってローダミンBを分解するが,λ>440 nm光照射下では分解速度が低下する。ここにUCPを添加すると,分解速度が向上した。すなわちUCPが照射光中の長波長光を短波長光(400~440 nm)に変換し,Pt/WO3光触媒を駆動させたと考えられる。
続いて,TTA-UCが水分解反応による酸素生成系に対しても利用可能であること実証するために,還元犠牲剤AgNO3存在中のMo: BiVO4光触媒による水の酸化反応と組み合わせた実験を行った33)。この実証では,Fig. 3(a)に示すTTA-UCメカニズムのうち,増感剤分子としてPt(OEP)を用い,DPAとともに粘土材料・合成サポナイト(Sumecton)層間に導入した。PtOEP-DPAの組み合わせでは,TTA-UCによって550 nm域の赤色光を400 nm域の青色光に変換できる。得られたUCP (DPA-PtOEP-Sumecton)をMo: BiVO4光触媒とともにAgNO3水溶液に分散し,520~600 nm Xeランプ照射によって生じる酸素量をFig. 4(b)にプロットした。Mo: BiVO4光触媒による酸素生成はUCPの添加によって有意に向上した(0.38→0.49 µmol h−1)。UCPが照射光中の600~520 nmに変換し,Mo: BiVO4光触媒を駆動させたと考えられる。
以上第3章では,粘土層間への発光性有機分子の集積によって,長波長光から短波長光への変換を可能にするフォトン・アップコンバージョン材料の開発について述べた。得られた材料を短波長光で駆動する光触媒とともに水に分散して長波長光を照射すると,フォトン・アップコンバージョンによって生じた短波長光によって光触媒を駆動させることができた。この成果の最も重要なポイントは,三重項励起状態をクエンチしてしまう水・酸素の存在下でもフォトン・アップコンバージョンが起こり,その光を使った触媒反応が進行できることである。
一方,無機ナノ空間でのTTA-UCの量子効率の低さ(~0.01%)が課題となる。特に,分子間で起こる励起三重項エネルギー移動過程が本系の全体の量子効率を支配していると考えられる。特に,疎水性相互作用を利用したDPAの層間導入量には限界があり,飽和濃度のDPA溶液を用いて層間導入を行ったとしても,有機元素分析から見積もられる層間のDPAの平均分子間距離は3.0 nmであり,励起エネルギー移動が起こるための条件としては遠い。十分な光吸収–発光が重要な要素となる発光体創出においては,ナノ空間に対する発光性有機分子の密な充填が重要である。続く第4章では,DPAのような無置換アリール分子を層間に密に充填させることのできる,新たな分子充填法の開発について述べる。
4. 細孔内脱アミン化法による層間への無置換アリール充填
嵩高い分子を,その分子と同等のサイズのナノ空間に大量に充填するためには,分子がナノ空間に入っていこうとするドライビングフォースが必要不可欠である。粘土材料やゼオライトのナノ空間は多くの場合,カチオン交換能を有しているため,静電相互作用をドライビングフォースとしたカチオン分子の充填が可能である。しかしながら,前項目のDPAのように,多くの興味深い発光特性を示す有機分子は中性・疎水性のアレーン誘導体であるため,ナノ空間への密な充填は難しい。
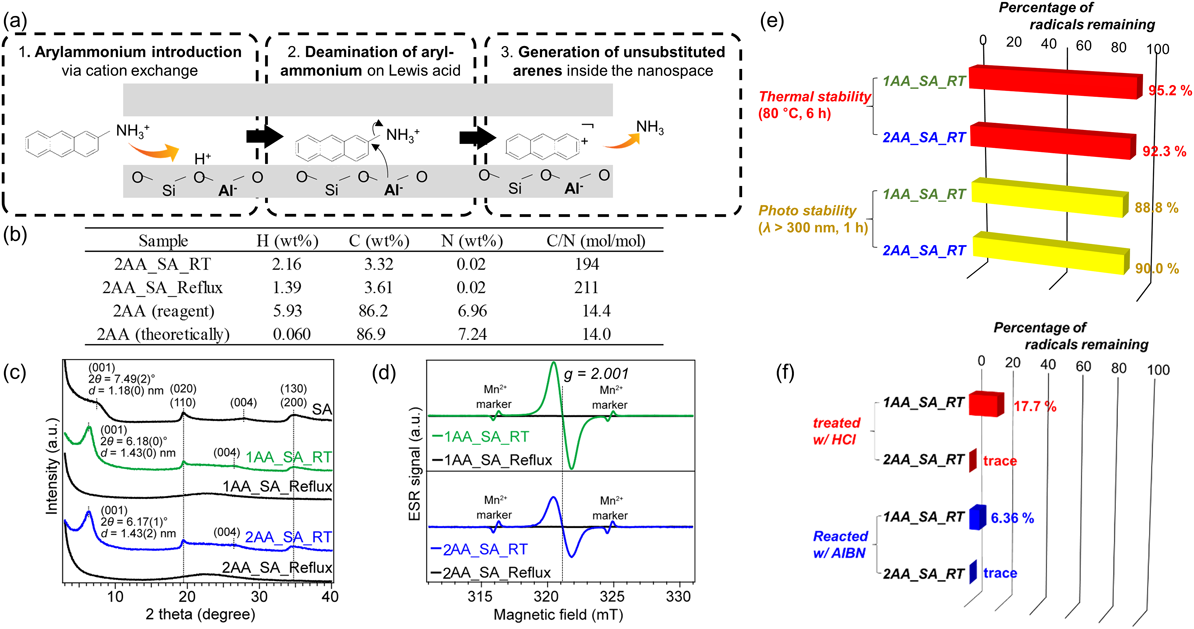
筆者は,アリールアンモニウムを粘土鉱物およびゼオライトのナノ空間にイオン交換法で導入すると,粘土・ゼオライトが有する触媒作用によってアンモニウム基の脱離が起こり,ナノ空間でアレーン分子が生成することを発見した(Fig. 5(a))34)。この方法は静電相互作用をドライビングフォースとしており,導入されたアリールアンモニウムのC-N結合をルイス酸点によって切断35)することで,ナノ空間に対して非常に密にアレーン分子を充填することができる。
4.1 サポナイト層間でのアリールアンモニウムの触媒的脱アミノ化による安定アリールラジカル生成
まず,粘土材料サポナイト層間に対するアレーン分子充填についての結果を示す。2-アミノアントラセンを50 mLのエタノールに溶解したのち,50 mLの1 M塩酸を加えた。この時,黄色を呈していた2-アミノアントラセン溶液が透明に変化したことから,2-アントリルアンモニウムの生成を確認した。この溶液に,クニミネ工業(株)から購入した合成サポナイト(SA)を500 mg分散して8時間室温もしくは還流条件で撹拌した。得られたサンプル(2AA_SA_RT or 2AA_SA_Reflux)のCHN元素分析によってサポナイトと2-アントリルアンモニウムの複合化を確認した(Fig. 5(b))。炭素成分はアントラセン環に由来するものであり,その量から導入されたアントラセン分子数を求めたところ0.184~0.290 mmol gsample−1となった。これは,SAのイオン交換容量の33.2~47.9%に相当する。一方で,本来アミノ基由来として含まれるはずの窒素はほとんど検出されなかったことから,2AAは脱アミノ化によって無置換のアントラセン環としてSAと複合化したと考えられる。
Fig. 5(c)に示したX線回折パターンから,アントラセン環がSA層間に存在することを確認した。原料SAの層状構造に対応するX線回折ピーク2θ=7.49°は,室温での2-アントリルアンモニウムとの反応によって2θ=6.18°に低角シフトした。すなわち,層間空間が増大したことから,アントラセン環が層間に導入されたと考えられる。更に,層間に導入されたアントラセン環はラジカル状態であることが,電子スピン共鳴(ESR)スペクトルから明らかとなった(Fig. 5(d))。2AA_SA_RTは有機物ラジカルに帰属されるg=2.001に強いピークを発現した。2,2,6,6-テトラメチルピペリジン1-オキシル(TEMPO)を用いた相対定量法によって,2AA_SA_RT中に導入されたアントラセンの40%程度がラジカル化していることが確認できた。更に,1-アントリルアンモニウムを出発物質として複合化した1AA_SA_RTでは,導入されたアントラセンのほぼ100%がラジカル化していることが分かった。
一方,還流条件で複合化した1AA_SA_Refluxおよび2AA_SA_Refluxではラジカルの存在を示すESRシグナルは観測されなかった。X線回折測定によって,1AA_SA_Refluxおよび2AA_SA_Refluxは層状構造が崩壊しアモルファス化していることが分かった。すなわち,アントラセンが層間ナノ空間に充填されていることで,ラジカル状態が安定化されて保持されることが明らかとなった。このラジカルは空気中でも非常に安定であり,1年以上空気中・暗所で放置したとしてもほとんどラジカル量が変化しないことが分かった。
この粘土層間アントラセンラジカルの特徴は,熱・光安定性も極めて高い点にある。80°C環境および紫外光照射下でもほとんどラジカルが失活しなかった(Fig. 5(e))。一方で,酸処理による層状粘土材料へのダメージによって,ラジカルは容易に失活した。また,AIBN熱分解によって生じるフリーラジカル存在下でもラジカル量が大幅に減少した(Fig. 5(f))。これらの事実は,層状構造がアントラセンラジカルを保護している一方で,層状構造の崩壊・剥離などの操作によって容易にラジカルを化学反応中に取り出せるという特長を示している。
以上の結果から,アントリルアンモニウムを出発物質としたナノ空間への無置換アントラセンの導入に成功し,粘土層間でこの無置換アントラセンはラジカル状態で保持されることが明らかとなった。その他のアリールアンモニウムとして,ナフチルアンモニウム,ピレンアンモニウムを用いても同様に対応するアリールラジカルが層間に充填されることも実証した。更に,他の置換基を有するナフチルアンモニウムに対しても同様に層間へのラジカル充填に成功した。すなわち,ナノ空間に充填したいアリール分子のアンモニウム置換型分子さえ用意できれば,ナノ空間内での脱アミノ化を介してアリール分子を密に充填することができる。
以降の節では,触媒的脱アミノ化を介してナノ空間に充填したアリール分子を用いた,光機能性材料の開発について,筆者らの最近の取り組みについて紹介する。
4.2 層間アリールラジカルを開始剤とする光機能性スチレンの創出
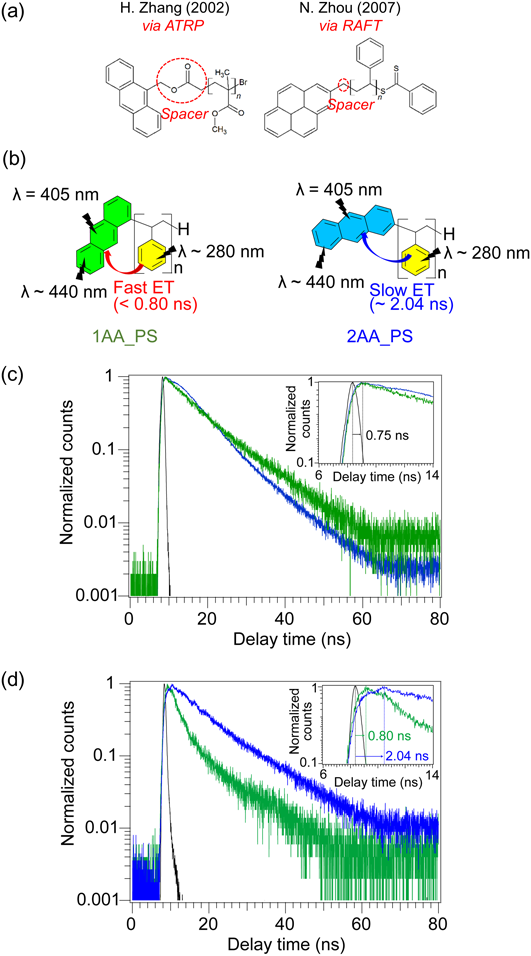
本節では,前節で得られた粘土層間で安定化された芳香族ラジカルを開始剤として,スチレンのラジカル重合を実証した36)。この方法で得られるポリスチレンは,高分子鎖末端に「直接」アリール基が結合した構造となる。近年,ドラックデリバリーや細胞イメージングなどに有用な,特有の末端基を有する機能性高分子材料が盛んに研究されている。例えばポリスチレン高分子末端へのアントラセン基配置は,原子移動ラジカル重合法(ATRP)37)や可逆的付加開裂連鎖移動重合法(RAFT)38)などの特殊な開始剤を用いた重合法によって達成されている。しかしながらこれらの重合法で得られるポリマーには,主鎖と末端基の間に開始剤由来のスペーサー部位が残ってしまい,「直接」末端基を結合することはできていない(Fig. 6(a))。また,これらの重合開始剤には不安定なものが多く,安全な保管に多くのコストを要する。
前節で得られたアントラセンラジカル含有サポナイトをラジカル開始剤としてスチレンを重合することで,アントラセンが末端に直接結合したポリスチレンを得た(Fig. 6(b))。紫外可視吸収光度計を検出器として用いたゲル浸透クロマトグラフィー法により,末端にアントラセンが結合したポリスチレンの数平均分子量は100,000程度となり,粘土層間のアントラセンラジカルが重合開始剤として機能したことを確認した。興味深いことに,アントラセン環へのポリスチレン主鎖の置換位置は,アントラセンラジカルの原料となるアントリルアンモニウムのアンモニウム基置換位置を反映していることが,Fig. 6(c),(d)に示す発光減衰測定の結果から示唆された。以下では,1-アントリルアンモニウムおよび2-アントリルアンモニウムを原料として得たポリスチレンをそれぞれ,1AA_PS,2AA_PSと表記する。
Fig. 6(c)の発光減衰は,1AA_PSおよび2AA_PSに対して波長405 nmの光を照射してアントラセン環を励起させ,アントラセンからの自己発光を波長440 nmで記録したものである。この時の発光減衰は,1AA_PSおよび2AA_PSでほとんど変化がないことから,アントラセン環の自己励起–発光挙動に大きな違いがないことが分かる。一方Fig. 6(d)では,波長280 nmの光を照射してポリスチレン鎖を励起させ,エネルギー移動を介して生じるアントラセン環からの発光を波長440 nmで記録した。この時,1AA_PSと比較して2AA_PSでは発光減衰の速度が遅いことが明らかとなった。更に励起光照射直後の発光挙動を詳細に分析すると,1AA_PSの発光極大は励起光照射から0.80 nsで生じるのに対して,2AA_PSでは発光極大が2.04 nsに現れた。これらの事実から,2AA_PS中で起こるポリスチレン鎖の光励起によるアントラセン環へのエネルギー移動が,1AA_PS中の同様のエネルギー移動と比較して遅いということが結論できる。このエネルギー移動ダイナミクスの違いは,ポリスチレン主鎖に対するアントラセン結合位置の違いを反映したエネルギー移動距離の違いに起因するものと考えられる。このような僅かな発光挙動の違いは,ポリスチレン主鎖に対してアントラセン環が直接結合した高分子材料を実現したからこそ,観測できる現象であることを主張したい。現在,これらのポリマーのNMRによる詳細な構造解析とともに,スチレン重合メカニズムの解析を進めている。
上記の方法と同様にして,ポリスチレン鎖末端へのナフタレン環,ピレン環の導入にも成功した。粘土層間へのアリールアンモニウムの導入によって生じるアリールラジカルを重合開始剤として,ポリスチレン鎖末端に「位置選択的に」アリール基を「直接結合」させることに初めて成功した例である。
4.3 ゼオライト細孔内への無置換アリールによるドラスティックな発光特性制御
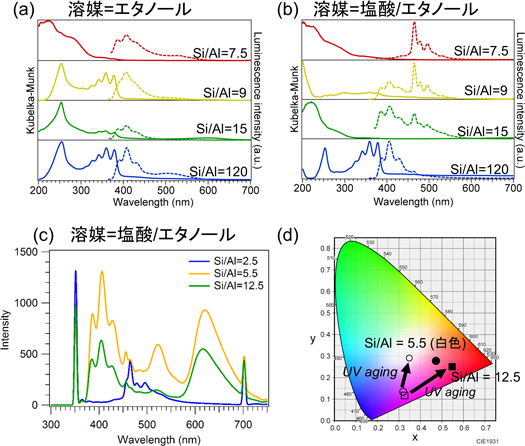
ゼオライトナノ空間に対しても,アントリルアンモニウムのイオン交換導入と,細孔内での触媒的脱アミノ化を介した無置換アントラセンの導入を試みた39)。エタノールもしくは塩酸/エタノールを溶媒に用いて2AAをH-MORゼオライトの細孔内に導入したところ,細孔内での脱アミン化によってアントラセンに転換されたことを元素分析などで確認した。H-MORゼオライトに導入されたアントラセンの発光スペクトル(Fig. 7(a)(b))のうち,塩酸/エタノール溶媒条件,Si/Al=7.5において極めて高い色純度の青色発光(半値全幅=8.57 nm@λ=465 nm)を観測した。有機色素の発光色純度が無機材料と比較して低いことが有機EL材料開発の一つの課題となっており,多くの有機合成化学的な方法による色純度の向上が試みられている40)。筆者の知る限り,今回の結果は有機発光体として最も高い色純度の青色発光である。
2AAをH-FAUゼオライトの細孔内に導入した場合も,脱アミン化によってアントラセンに転換された。H-FAU内のアントラセンは,紫外光(λ=370 nm)の照射によって発光挙動が変化した。十分な時間の紫外光照射後,発光スペクトルを測定した結果をFig. 7(c)に示す。Si/Al=5.5のFAUに導入したアントラセンは,400,510,620 nm近傍に,それぞれ発光ピークを示した。これらの発光波長はそれぞれ青・緑・赤に対応しており,この発光スペクトルは白色光に対応することが分かった(Fig. 7(d))。白色発光体も,低コストな有機EL材料創出に向けて重要なキーテーマである。おそらくアントラセン分子単一での白色発光を実証した例は他にない。現在,発光色変化のメカニズム究明を進めている。
我々は,層状材料の層間ナノ空間,およびゼオライトの細孔ナノ空間への発光性分子充填を軸に,エネルギー問題および最先端光デバイス・センサーに資する光機能性材料を開発してきた。光エネルギー有効利用を目指して,光触媒ナノシート積層構造の精密設計やナノ空間への分子配置などによる光誘起電子移動・エネルギー移動の制御を実証した。近年の人工光合成システム開発の勢いは非常に強く,社会実装目前のところまで来ている。ナノ空間を使った光エネルギー変換を意義ある研究とするためには,質的な光化学過程の制御のみならず,量的なエネルギー転換の実現が求められるだろう。この観点から,十分な光吸収とエネルギー転換を起こすためにはナノ空間への密な分子充填を実現する必要がある。従来のナノ空間への分子導入方法は,イオン交換法以外に密な分子充填を実現する方法がなかった。筆者らは,中性発光材料群のナノ空間への充填を目指して,イオン交換導入したアリールアンモニウムのナノ空間内での触媒的脱アミノ化による無置換アリール生成による分子充填法を開発した。この分子充填法を使うことで,これまで困難だった嵩高い分子の狭いナノ空間への導入も期待でき,新たなホスト–ゲスト化学による光機能性材料の開発が拓けるであろう。
謝辞Acknowledgments
今回の解説記事に記載した研究を遂行するにあたり,東京大学・化学システム工学専攻,大久保達也教授,脇原徹教授には,私を日本学術振興会特別研究員SPDとして受け入れていただき,無機多孔体合成およびナノ空間化学に関する有益な助言をいただきました。また,東京工業大学・科学技術創成研究院,和田雄二特任教授・名誉教授および東京電機大学・応用化学科,望月大准教授には半導体ナノシート合成・設計と光化学分析に関して,博士論文研究を通して熱心にご指導いただきました。この場を借りて各先生方に御礼申し上げます。
引用文献References
1) M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis, A. W. Ho-Baillie, Prog. Photovolt. Res. Appl., 28, 3 (2020).
2) S. Ardo, D. F. Rivas, M. A. Modestino, V. S. Greiving, F. F. Abdi, E. A. Llado, V. Artero, K. Ayers, C. Battaglia, J.-P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. D. Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J.-W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg, P. Westerik, Energy Environ. Sci., 11, 2768(2018).
3) N. S. Lewis, Science, 351, 353(2016).
4) A. Fujishima, K. Honda, Nature, 238, 37(1972).
5) T. Higashi, H. Nishiyama, Y. Suzuki, Y. Sasaki, T. Hisatomi, M. Katayama, T. Minegishi, K. Seki, T. Yamada, K. Domen, Angew. Chem. Int. Ed., 58, 2300(2019).
6) W. H. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater, H. J. Lewerenz, ACS Energy Lett., 3, 1795(2018).
7) Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada, K. Domen, Nature Mater., 15, 611(2016).
8) K. Takanabe, ACS Catal., 7, 8006(2017).
9) T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi, K. Domen, Nature, 581, 411(2020).
10) C. Adachi, Jpn. J. Appl. Phys., 53, 060101(2014).
11) S. Ohta, D. Glancy, W. C. Chan, Science, 351, 841(2016).
12) Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook, J. Y. Lee, Adv. Funct. Mater., 27, 1603007(2017).
13) G. D. Stucky, J. E. Mac Dougall, Science, 247, 669(1990).
14) G. Schulz-Ekloff, D. Wöhrle, B. van Duffel, R. A. Schoonheydt, Microporous and Mesoporous Mater., 51, 91(2002).
15) Y. Wada, C. Nakaoka, A. Morikawa, Chem. Lett., 17, 25(1988).
16) Y. Wada, Y. Yoshizawa, A. Morikawa, J. Chem. Soc. Chem. Commun., 4, 319(1990).
17) Y. Wada, T. Okubo, M. Ryo, T. Nakazawa, Y. Hasegawa, S. Yanagida, J. Am. Chem. Soc., 122, 8583(2000).
18) S. Hashimoto, S. Ikuta, T. Asahi, H. Masuhara, Langmuir, 14, 4284(1998).
19) F. Márquez, H. García, E. Palomares, L. Fernández, A. Corma, J. Am. Chem. Soc., 122, 6520(2000).
20) H. Vezin, A. Moissette, C. Brémard, Angew. Chem. Int. Ed., 42, 5587(2003).
21) U. Vietze, O. Krauss, F. Laeri, G. Ihlein, F. Schüth, B. Limburg, M. Abraham, Phys. Rev. Lett., 81, 4628(1998).
22) F. Marlow, M. D. McGehee, D. Zhao, B. F. Chmelka, G. D. Stucky, Adv. Mater., 11, 632(1999).
23) S. Megelski, A. Lieb, M. Pauchard, A. Drechsler, S. Glaus, C. Debus, A. J. Meixner, G. Calzaferri, J. Phys. Chem. B, 105, 25(2001).
24) R. Abe, J. Photochem. Photobiol. C, 11, 179(2010).
25) D. Mochizuki, K. Kumagai, M. M. Maitani, Y. Wada, Angew. Chem. Int. Ed., 51, 5452(2012).
26) F. Kishimoto, T. Ano, D. Mochizuki, T. Terauchi, M. M. Maitani, E. Suzuki, Y. Wada, RSC Adv., 6, 73830(2016).
27) F. Kishimoto, D. Mochizuki, K. Kumagai, M. M. Maitani, E. Suzuki, Y. Wada, Phys. Chem. Chem. Phys., 16, 872(2014).
28) F. Kishimoto, D. Mochizuki, M. M. Maitani, E. Suzuki, Y. Wada, ACS Appl. Mater. Interfaces, 10, 37150(2018).
29) T. Ano, F. Kishimoto, D. Mochizuki, S. Tsubaki, M. M. Maitani, E. Suzuki, Y. Wada, Chem. Lett., 45, 1111(2016).
30) F. Kishimoto, D. Mochizuki, M. M. Maitani, E. Suzuki, Y. Wada, J. Phys. Chem. C, 123, 30029(2019).
31) D. Mochizuki, K. Kumagai, M. M. Maitani, E. Suzuki, Y. Wada, J. Phys. Chem. C, 118, 22968(2014).
32) F. Kishimoto, T. Wakihara, T. Okubo, ACS Appl. Mater. Interfaces, 12, 7021(2020).
33) Y. Kageshima, S. Tateyama, F. Kishimoto, K. Teshima, K. Domen, H Nishikiori, Phys. Chem. Chem. Phys., 23, 5673(2021).
34) F. Kishimoto, K. Hisano, T. Wakihara, T. Okubo, Chem. Mater., 32, 9008(2020).
35) Z. X. Wang, B. Yang, Org. Biomol. Chem., 18, 1057(2020).
36) F. Kishimoto, K. Hisano, T. Wakihara, T. Okubo, Dalton Trans., 50, 835(2021).
37) H. Zhang, B. Klumperman, R. van der Linde, Macromolecules, 35, 2261(2002).
38) N. Zhou, L. Lu, J. Zhu, X. Yang, X. Wang, X. Zhu, Z. Zhang, Polymer, 48, 1255(2007).
39) 岸本史直,久野恭平,堤 治,脇原 徹,大久保達也,第35回ゼオライト研究発表会,C25総(2019).
40) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai, T. Hatakeyama, Nature Photonics, 13, 678(2019).