両親媒性有機分子を利用して合成する規則性多孔体の研究分野では,シリカやカーボンといった組成を除くと,溶媒揮発法による薄膜の合成例がほとんどである1)。溶媒揮発法とは,例えば,透明な前駆溶液を基板上にコーティングすることでメソポーラス薄膜を簡便に得る方法のことを指す。コーティング直後に溶媒(主にエタノール)が速やかに揮発するので,前駆溶液の濃度が高くなった結果として,両親媒性有機分子の自己集合が誘起される。その過程で,前駆溶液中の溶解種のネットワーク形成が適切に調節できた場合のみ,構造規則性が高いメソポーラス薄膜を得ることができる。溶媒揮発法を粉体合成に適用した例もいくつか報告されているが,揮発させなければならない溶媒量が多くなってしまうので,前駆溶液中での反応性を制御することが難しい無機組成の場合は特に,規則的なメソポーラス構造を有する粉体試料を十分に提供できる状況にないのが現状である。
メソポーラスアルミナ粉体の合成例を紹介する2–4)。いずれの場合も,酸性条件下でポリオキシエチレン(EO)とポリオキシプロピレン(PO)からなるトリブロック共重合体(EOnPOmEOn)を含む均質な前駆溶液を調製したものしかない5)。従って,精密な孔径制御の可能性や異なる構造規則性の設計等には至っておらず,両親媒性有機分子が溶液中で自己集合する性質を利用する合成法の特徴は十分に活かされていない。ただし,メソポーラス構造を保持したままアルミナ骨格をγ相にある程度結晶化できることも示されていたため3),高比表面積の触媒担体として広く利用できると思われた。しかしながら,最初の報告から10年以上が経過した現在も,種々の構造規則性を有するメソポーラスアルミナを利用した応用研究の報告例を見ることは多くない4)。我々も数種類のEOnPOmEOnを用いて追試を試み,ある程度は多孔化できることまでは確認したが,報告例にあるようなドライアッププロセスによる構造規則性の高いメソポーラスアルミナの合成は未だ実現できていない。
以上の背景から,再現良く前駆溶液から溶媒を除去,乾燥し,粉体試料が回収できる合成プロセスの設計に主眼を置き,多孔質アルミナの粉体合成技術の開発を開始した。ここ数年の検討で,比表面積の大きいメソポーラスアルミナの粉体合成に成功するとともに,その実現に必要な構造モデルを提案することもできた6,7)。また,非対称のブロック共重合体(ポリスチレンとポリオキシエチレンの共重合体,PS-b-PEO)を利用した孔径増大にも取り組み,その結果,アルミナ骨格の結晶化挙動の理解を深めることもできている8,9)。本解説では,両親媒性有機分子を利用したメソポーラスアルミナの粉体合成に関して,高表面積化と高結晶化の実現に向けた取り組みを中心に紹介する。
2. 両親媒性有機分子を利用した規則性多孔体の合成
アルキルトリメチルアンモニウム(CnTMA)やEOnPOmEOn等の分子集合体を鋳型として,材料内部にナノメートルレベルの周期構造を設計する合成法のことを超分子鋳型法と呼ぶ。代表的なものには,CnTMAやEOnPOmEOnを用いて合成されたメソポーラスシリカ(MCM-41,SBA-15等)がある10,11)。しかし,超分子鋳型法で合成されるメソポーラス材料の場合,孔径が大きくなると酸化物骨格の厚みが増し,比表面積は小さくなる。そのため,触媒担体等への展開を想定する場合は,性能向上と直結する材料表面の活性サイト数と孔径で決まる反応物の拡散性を最適化するように構造設計していく必要がある。加えて,酸化物材料に結晶性が求められる用途では,結晶化に伴う比表面積の低下を抑制する精密な多孔化技術が求められる。本当の意味で機能性メソポーラス材料を合成するという見地から,我々は,多孔性を保持したまま酸化物骨格の結晶性を最大化するような高度な超分子鋳型法の開発を目指している。
一般的なメソポーラスシリカの粉体合成の場合は,溶解ケイ酸種が両親媒性有機分子と相互作用し,ケイ酸骨格が徐々に形成される過程で,有機分子集合体を取り囲むようにケイ酸種が高分子量化して沈殿(粉体試料)が回収される10,11)。両親媒性有機分子を利用したメソポーラスアルミナの初期の合成例は,本紙に掲載あるいは筆者の解説記事12,13)を参照されたいが,最近の報告3)では,高比表面積(約400 m2 g−1)のγ-アルミナ粉末の合成の可能性について言及されており,他の研究グループからも類似材料が報告されている4)。我々がこれらの合成を再現できていないことは前述したが,その困難さの根拠は,前駆溶液をトレイに移して溶媒除去するドライアッププロセスが含まれているためではないかと考えている。すなわち,溶媒の揮発速度がトレイの大きさ(液面の面積)や実験室の環境(温度や湿度)等の条件に依存してしまうため,その実験を忠実に再現することができていないと考察している。
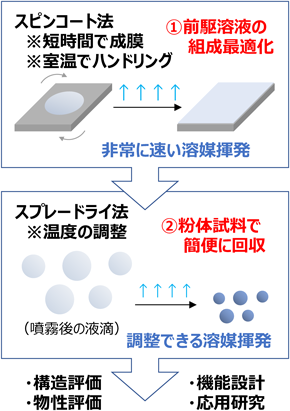
以上の経緯から,合成条件が再現できないドライアッププロセス2–4)の再検証を継続するよりもむしろ,メソポーラスアルミナの新しい合成法を開発した方が建設的であると判断した。また,我々は,成膜プロセスを粉体合成に拡張するための方法論(図1参照)として,噴霧乾燥プロセスを活用した。メソポーラス材料の薄膜化では,透明で均質な前駆溶液を調製し,その前駆溶液をスピンコート法等で基板上に成膜する。薄く広げた前駆溶液からは溶媒が短時間で揮発するため,前駆溶液中での無機種の反応が必要以上に進行する前に,前駆溶液が濃縮されて両親媒性有機分子の自己集合が完了する。従って,メソポーラス薄膜の合成用に最適化した透明な前駆溶液を適切な温度で噴霧乾燥するだけで,速やかに多孔質構造を有する粉体試料が回収できると考えた。そして,前駆溶液の組成最適化が成膜プロセスだけで迅速に完結でき,粉体合成を実現するまでの作業時間が大幅に短縮されることも期待した。
3. 両親媒性有機分子を利用した多孔質アルミナの粉体合成
3.1 高表面積化を実現する構造モデルの提案
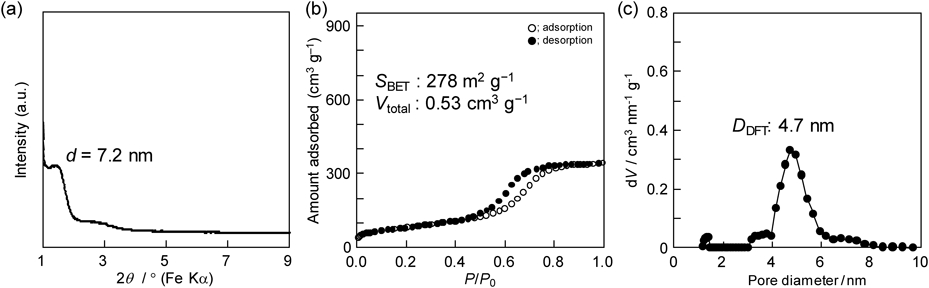
構造規則性が高いメソポーラスアルミナ薄膜の報告例14,15)を参考に,前駆溶液を調製する条件を再検証し,噴霧乾燥により粉体試料を合成した6)。濃塩酸を含むエタノール(EtOH)溶液に,激しく攪拌しながら,アルミニウムn-ブトキシド(Al(On-C4H9)3)を添加した。所定の時間,アルミニウム源の加水分解反応を進行させた後に,EOnPOmEOn系の両親媒性有機分子(例えば,Pluronic P123:EO20PO70EO20)を溶解したEtOH溶液と混合し,透明な前駆溶液を得た。調製した前駆溶液を噴霧乾燥(170°C)することで粉体試料を回収した。構造規則性や多孔性を評価するために,400°C焼成後の試料について,粉末X線回折(XRD)測定,窒素吸着測定等の各種分析を行った(図2参照)。例えば,窒素吸着測定から,平均直径4.7 nmの比較的サイズの揃ったメソ孔の存在を確認した。比表面積は280 m2 g−1程度と比較的低い値だったが,噴霧乾燥で得られる粉体試料の比表面積は小さくなる傾向があり16),我々は,その原因は,噴霧乾燥に特徴的な球状粒子が生成する過程で均一メソ孔が幾何学的に最密充填できないためと予想した(図3上段参照)。なお,透過型電子顕微鏡(TEM)観察から,粒子全体に均一なメソ孔が導入されたように見えるが,窒素吸着測定により算出した多孔性も十分に考慮し,粒子全体にメソ孔が導入されたかを結論する必要がある。
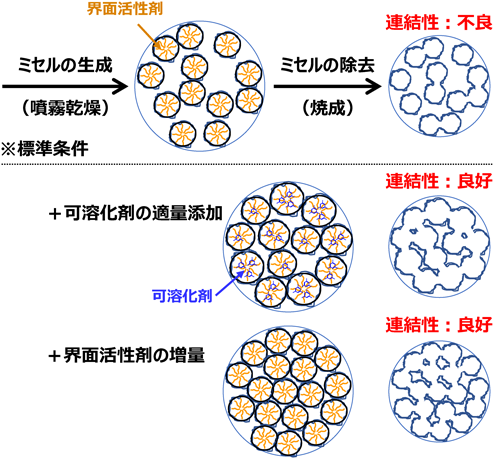
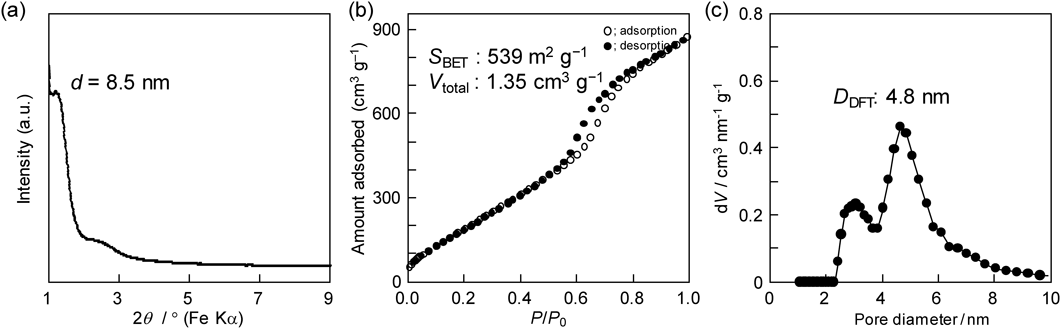
他方,EO20PO70EO20を用いて合成したメソポーラスアルミナの平均孔径が5 nm未満と見積られたため,孔径の増大によるターゲット分子の拡散性の向上を目指し,可溶化剤として機能することが知られている1,3,5-トリメチルベンゼン(1,3,5-TMB)および1,3,5-トリイソプロピルベンゼン(1,3,5-TIPBz)を添加した合成も行った6)。種々の検討を重ねた結果,例えば1,3,5-TIPBzを適量添加すると,比表面積が2倍程度(約540 m2 g−1)に増大することを見出した(図4参照)。特徴的な結果だけを説明すると,孔径分布曲線から明らかなように,孔径約3 nmに相当する新たなメソ孔の生成が確認されている。有機助剤の添加が球状メソ孔を僅かに増大させ,隣接する球状メソ孔の距離が縮まった結果,新たな連結孔が形成すると考察した(図3中段参照)。また,この結論は,界面活性剤の添加量を増やすだけでも球状ミセルの数が増え,隣接する距離が縮まり,球状メソ孔の連結性が改善,比表面積が500 m2 g−1程度にまで増大できたこと(図3下段参照)からも裏付けられている7)。
3.2 大孔径化への取り組みとアルミナ骨格の結晶化挙動の理解
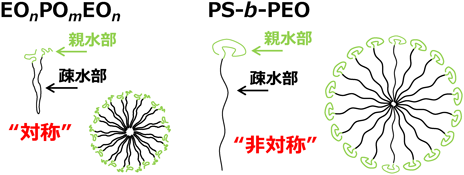
前章で説明した通り,EO20PO70EO20を利用した合成では,可溶化剤を添加しても孔径の顕著な増大が認められなかったため,より大きな自己集合体を形成する非対称型のジブロック共重合体(例えば,ポリスチレン–ポリオキシエチレンジブロック共重合体:PS-b-PEO)を用いた合成法も検討した8,9)。溶液中でのEO20PO70EO20の自己集合挙動を考えると,親水部であるEOユニットがシェル構造になるようミセルを形成させるためには,疎水部であるPOユニットが折れ曲がらなければならない(図5左参照)。非対称の分子構造を有するPS-b-PEOがミセルを形成する場合は,疎水部が折れ曲がる必要がないため,より大きなミセルを形成すると考えられる(図5右参照)。
この場合も,PS-b-PEOを用いた規則性の高い多孔質アルミナ薄膜の報告例があったため17),その追試から開始し,前駆溶液の組成最適化が終了したと判断した時点で噴霧乾燥プロセスへと適用した。使用したPS-b-PEOの各ブロックの分子量で表記するが,35000-b-17000を用いた場合を例に説明すると9),孔径を均質化するためにテトラヒドロフラン(THF)/EtOH比を最適化した後に,アルミニウム源である塩化アルミニウム(AlCl3)量を減らす等した結果,噴霧乾燥によって比較的均質なナノ空間が導入されたアルミナ粉体を回収できた。例えば,粒子表面全体にナノ空間が存在している様子を走査型電子顕微鏡(SEM)により観察し,TEM観察で平均直径が40 nm程度の球状空間が粒子全体に存在していることを確認した(図6参照)。SEM観察からも孔径を類推することは可能だが,SEM観察で孔径(=最大直径)を正確に評価することは簡単ではなく,参考程度の数値と考えることをお勧めする。また,孔径の増大に伴いアルミナ骨格の厚みが増した結果,比表面積は240 m2 g−1程度となったが,孔径の増大によりナノ空間の容積は1.27 cm3 g−1にも達した。加えて,アルミナ骨格の内部にPEOユニットが挿入されて小さなメソ孔が形成することもあるため,比表面積の解釈にも十分な注意が必要だろう。
PS-b-PEOの分子集合体を利用したアルミナの多孔化に関しては,孔径はPSユニットの分子量に支配されやすい傾向が見られており,例えば,58500-b-37000を用いた合成では,孔径200 nm前後(SEMとTEMにより総合的に評価)の多孔質アルミナが得られた(図7参照)8)。従って,前駆溶液中で均質に溶解するような適切なPSユニットとPEOユニットの比となっている分子構造のPS-b-PEOを用いれば,PSユニットの分子量に依存した孔径制御が実現できることを意味している。他方,前述の35000-b-17000を用いた多孔質アルミナの合成でも,可溶化剤としてポリスチレンのホモポリマー(h-PS)を適量添加することで,その孔径が1.5倍程度にまで増大することも確認したため9),これらの手法を組み合わせれば,PSユニットの分子量に依存した段階的な孔径制御とh-PSの添加量による連続的な孔径増大も実現できるだろう。
3.3 アルミナ骨格の結晶化挙動の理解
孔径の異なる数種類の多孔質アルミナの粉体試料が合成できたため,多孔質アルミナ前駆物質を400°Cから1000°C(昇温速度:2°C min−1)の範囲で焼成し,表面曲率や厚みの異なるアルミナ骨格の結晶化挙動とナノメートルレベルの構造規則性や多孔性との相関を調査した6,8,9)。また,アルミナ骨格を高温相であるα相に転移させるために1300°Cでも焼成した。高角度領域のXRD測定で焼成過程に於ける酸化物骨格の結晶化挙動と結晶構造を同定した。また,TEM観察により多孔質構造の存在を確認するとともに,XRD測定,SEM観察,並びに窒素吸着測定も行い,両親媒性有機分子の自己集合により付与した多孔質構造に関する情報や焼成温度と多孔性との相関は総合的に判断した。
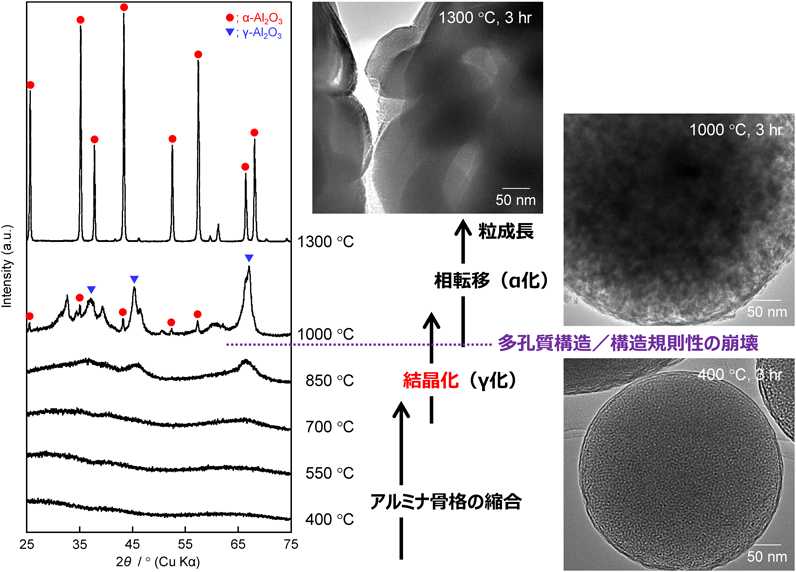
EO20PO70EO20を用いて合成したメソポーラスアルミナ(孔径:約5 nm)の場合から説明していく。例えば,1,3,5-TMBを添加した場合に得られた試料の場合(図8参照),700°C焼成した試料のアルミナ骨格はまだ非晶質だが,850°C焼成した試料ではγ相への結晶化が開始していた。XRD測定により,比較的高温(例えば850°C)で焼成した試料でも,低角度領域に回折ピークが存在しており,多孔性(構造規則性)を保持したままある程度は結晶化できることを確認した。ただし,それ以上の焼成温度,例えば1000°Cでは,アルミナ骨格のγ相への結晶化度は高くなったが,構造規則性は完全に崩壊していた。構造規則性が崩壊した後は,α相への相転移も確認された。焼成温度の上昇に伴い,アルミナ骨格が収縮,更にはγ相へと結晶化するため,アルミナ骨格の密度が増して比表面積は徐々に減少した。例えば,多孔質構造が保持されていた850°C焼成した試料の比表面積は300 m2 g−1前後の値を維持したが,1000°C焼成した試料では,多孔質構造が崩壊した結果,比表面積も50 m2 g−1程度にまで大きく減少していた。
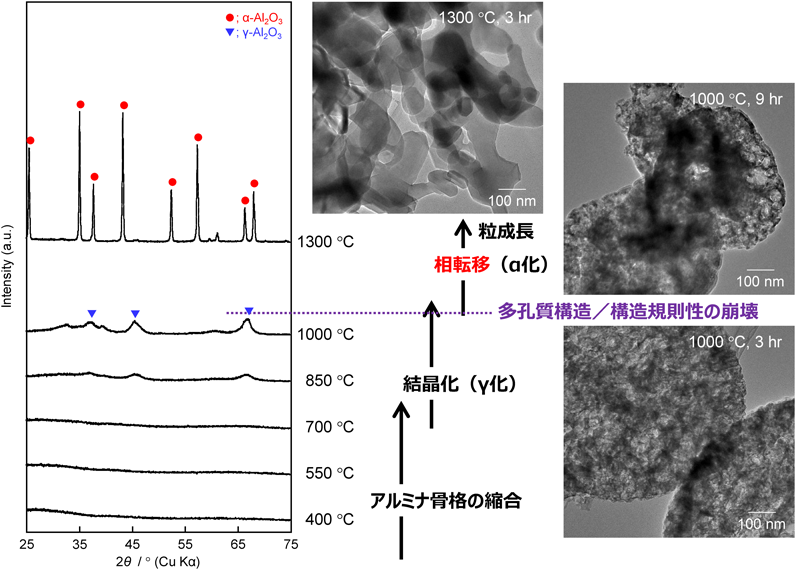
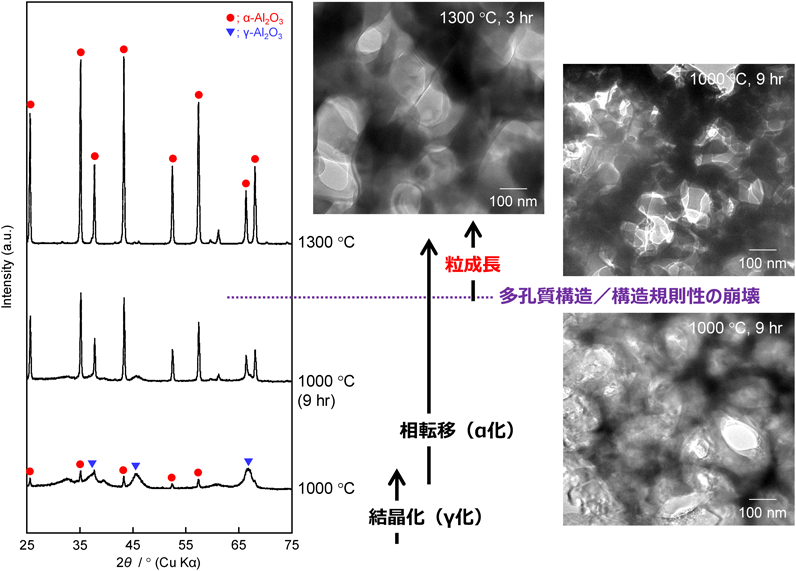
次に35000-b-17000を用いて合成した多孔質アルミナ(孔径:約40 nm)の場合(図9参照)を説明する。アルミナ骨格が厚くなったため,焼成温度を高くしても,少なくとも1000°Cまでは,比表面積は徐々に減少するだけだった9)。35000-b-17000の自己集合体が形成されたことに由来する多孔質構造に着目してTEM観察を行うと,焼成温度の上昇とともに,アルミナ骨格が徐々に収縮し,それに伴って平均孔径は小さくなった。例えば,1000°C焼成後,平均孔径が20 nm前後にまで収縮していた。ただし,焼成時間が3時間程度では,多孔質構造の崩壊はそれほど進行せず,α相への相転移もほとんど確認されることはなかった。しかし,焼成時間を延長すると多孔質構造が崩壊している箇所も混在し始めた。従って,平均孔径が40 nm程度の多孔質アルミナに関しては,1000°C程度がγ相の結晶化度を最大にする境界温度であると推察される。また,孔径分布等を丁寧に確認しながら,比表面積の変化についても議論した結果,850°Cより高温で焼成した試料では,PEOユニットが挿入されて生成した小さなメソ孔がアルミナ骨格の収縮によってほぼ消失し,850°Cあるいは1000°Cで焼成した多孔質アルミナ粉体の比表面積はそれぞれ165 m2 g−1および約95 m2 g−1と見積もられた。
以上の結晶化挙動は,アルミナ骨格の表面曲率の相違で説明できる8)。アルミナ骨格の表面曲率が大きい(孔径が小さい)と収縮等によりアルミナ骨格の表面近傍の密度がより高い状態になると考えられる。この制約が,孔径が小さい場合に結晶化が阻害される要因となる6)。孔径が小さい場合でも,高温で焼成した後に構造規則性が崩壊すれば,この制約がなくなるため,より結晶化しやすい環境へと変化する(図8参照)。ナノメートルレベルの構造規則性が崩壊した後に,γ相の結晶化度が大きくなったこと,更には部分的にα相への相転移が確認されたこととも関係がある。逆に,アルミナ骨格の表面曲率が小さい(孔径が大きい)とこの制約は緩和されると考えるのが妥当である9)。ただし,多孔質構造が保持されている限りは,アルミナ骨格の内部のみで結晶化が進行するため,その厚みによって結晶成長あるいは相転移が制限される。その結果,平均孔径が40 nm程度の多孔質アルミナの場合,焼成温度の上昇とともにγ相の結晶化度は増大したが,1000°C焼成でもα相への相転移が制限されたのだろう(図9参照)。
アルミナ骨格の表面曲率を用いた結晶化挙動の理解を拡張すると,例えば,更に大きな直径のナノ空間を有する多孔質アルミナであれば,多孔質構造を保持したままでより結晶化度の高い多孔質アルミナを得ることができるということになる。そこで,孔径が200 nm前後の多孔質アルミナに関しては,高温域を中心に,その結晶化挙動を調査した(図10参照)。表面曲率が非常に小さいため,予想通り,多孔質構造を保持したまま,アルミナ骨格のγ化だけでなく,α相への相転移が実現できることを見出した8)。アルミナ骨格が更に厚くなっていることを反映して,この場合は,1000°C焼成で,多孔質構造を保持したまま,α相への相転移が開始していた。焼成時間を延長すると,多孔質構造を保持したままで,α化が更に進行し,現時点では,α化率60%かつ比表面積が30 m2 g−1超の多孔質アルミナの合成に成功している。以上,表面曲率(≒孔径)とアルミナ骨格の結晶化挙動の関係が明らかになりつつある。種々の孔径を有するより多くの多孔質アルミナが合成できてくれば,より詳細な結晶化挙動としての理解が深まり,各種多孔質酸化物の設計指針となるだろう。