ゼオライトは触媒,吸着剤,イオン交換剤等として石油化学,化学分野をはじめ,幅広く使用されている。その理由として,ゼオライトが有する均一な分子レベルの細孔による分子ふるい能はもちろんのこと,骨格中のSi/Alにより親疎水性,吸着性,触媒活性を制御できることが挙げられる。さらには,メソ孔の導入や結晶形態制御による分子拡散促進1,2),結晶内Al導入部位制御による反応選択性の制御3),分離膜における配向性制御による分離選択性の向上4)等,高機能化手法が報告されている。このようなゼオライトの高機能化手法のなかで,吸着性能向上に効果的な手法として表面修飾(改質)が知られており,クロロシラン修飾による水中VOC除去5),界面活性剤やキトサン修飾による硝酸イオン除去6),アミノシラン修飾による色素回収7),シリカ修飾による吸着速度の向上8)等が報告されている。
表面修飾による高機能化は触媒への応用においても効果的であり,特に表面修飾ZSM-5を用いたパラキシレンの選択的合成は盛んに研究されてきた。古くはリン,ホウ素,MgOなどによる修飾9,10)が,また,1980年代中頃にCVDやCLDによるシリカ被覆ZSM-5が優れたパラキシレン選択性を示すことが報告されて以来11),本手法を用いた研究が数多く報告されている12,13)。近年では,メカノケミカル法による表面改質が選択性向上に効果的であるとの報告14)もあり,ゼオライトの表面修飾は触媒分野における高機能化手法のひとつとして今後も重要な技術と言える。筆者らはこれまでにZSM-5の外表面を純シリカMFI型ゼオライトであるsilicalite-1で被覆したコアシェル構造化ゼオライトがトルエンのアルキル化において極めて優れたパラキシレン選択性を示すことを見いだしており15),いくつかの研究グループが同様の成果を発表している16–18)。
本触媒で用いたZSM-5, silicalite-1ともMFI型の結晶構造を有しており,shell層となるsilicalite-1はZSM-5上にエピタキシャル成長することがわかっている19)。そのため,silicalite-1被覆による分子の細孔内拡散への影響は軽微であると予想されることから,これら生成物選択性の向上は,主として結晶外表面の不活性化による副反応の抑制に起因するものと考えてきた。しかしながら,silicalite-1被覆が細孔内拡散および分子ふるい能に及ぼす影響については不明のままであった。本稿では,MFI型ゼオライトを中心に,コアシェル構造化が吸着性能に及ぼす影響を概説するとともに,コアシェル構造化がゼオライトの吸着性能向上の汎用的な手法となり得ることを紹介する。
2. コアシェル構造化MFI型ゼオライトの吸着特性
2.1 MFI型ゼオライトのコアシェル構造化20)
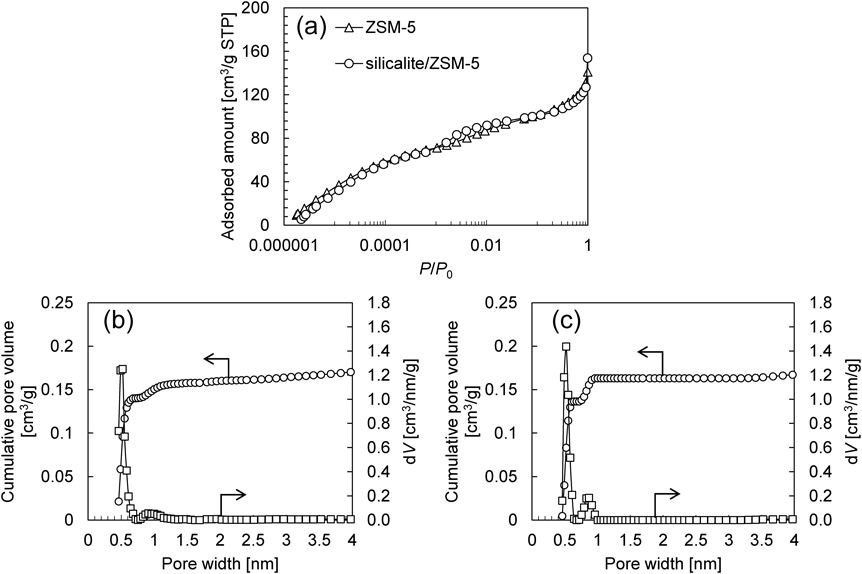
ゼオライトのコアシェル構造化はcoreとなるゼオライトの二次成長により可能であり,合成条件に若干の差異はあるものの,通常の水熱合成法が用いられる。Fig. 1にZSM-5および既報21)を参考にフッ化物法を用いたコアシェル構造化MFI型ゼオライト(silicalite/ZSM-5)のSEM像を示す。Silicalite/ZSM-5の外観からはcoreであるZSM-5が二次成長している様子が伺え,フッ化物法を用いた場合でも従来法と同様にsilicalire-1がZSM-5上にエピタキシャル成長していると考えられる。また,SEM像から見積もったZSM-5およびsilicalite/ZSM-5の平均結晶径はそれぞれ2.65 µmおよび2.77 µmであったことから,silicalite-1層の厚みは50 nm程度と予想される。Fig. 2に示すようにAr吸着等温線はいずれもI型を示しており,NLDFTにより求めたZSM-5およびsilicalite/ZSM-5それぞれのミクロ孔容積は0.160 cm3および0.163 cm3とほぼ同程度であった。細孔径分布を見ると,メインピークはMFIの細孔由来でありサンプル間に大きな違いは認められない。一方で9 Å付近に確認できるピークはコアシェル構造化により大きくなった。このピークはMFIのミクロ孔構造に直接関係はないものの,そのピーク強度は骨格内Alの減少により強くなることが知られている22)。つまり,この結果はsilicalite/ZSM-5のSi/AlがZSM-5に比べ大きいことを意味しており,XRFによる組成分析結果(Table 1)と一致する。これらの結果は,純シリカ組成のsilicalite-1層がZSM-5上に形成されていることを示唆するものであり,特にAr吸着等温線はコアシェル構造化がゼオライトのミクロ孔構造に影響していないことを示している。
Table 1. Physical properties of ZSM-5 and silicalite/ZSM-5 | Si/Ala | Crystal size D50 [µm] | Micropore volume [cm3/g]b | Total pore volume [cm3/g] |
|---|
| ZSM-5 | 34.4 | 2.65 | 0.160 | 0.224 |
| Silicalite/ZSM-5 | 55.0 | 2.77 | 0.163 | 0.242 |
| ameasured by XRF, bcalculated by NLDFT method |
2.2 キシレン異性体のクロマトグラフィー分離20)
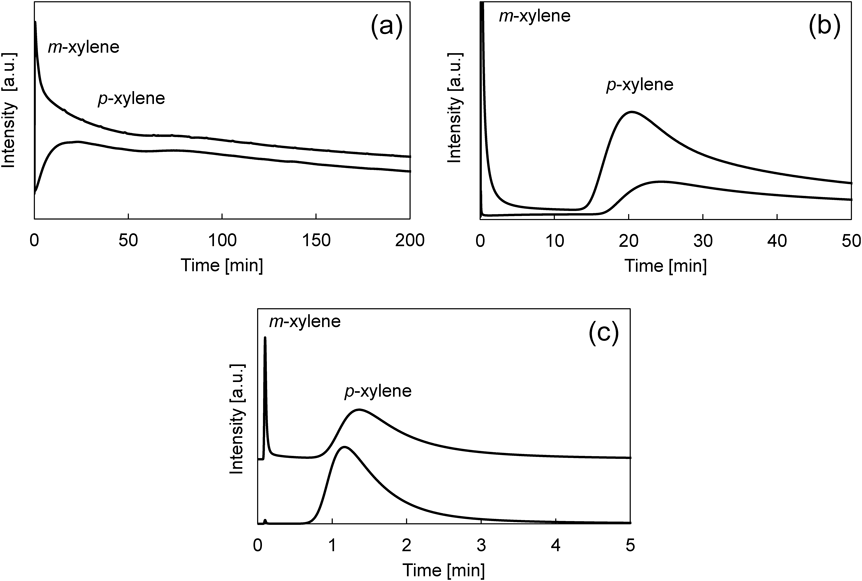
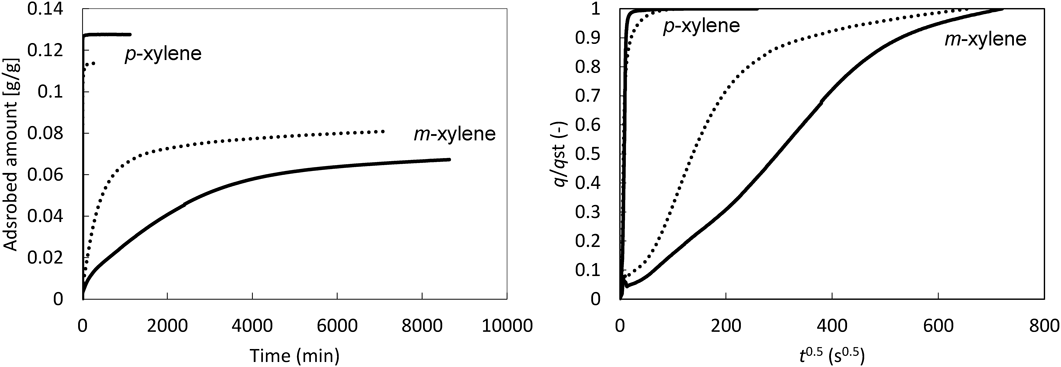
ZSM-5およびsilicalite/ZSM-5それぞれを充填したカラムを作製し,キシレン異性体(パラキシレン/メタキシレン)の分離性能を評価した。Fig. 3にZSM-5, silicalite/ZSM-5およびsilicalite-1のクロマトグラムを示す。なお,silicalite-1はコアシェル構造化の際の合成条件を用い,coreとなるZSM-5を添加せずに合成したものである。ZSM-5を用いた場合,メタキシレンの保持時間は非常に小さいにもかかわらず,メタキシレンのピークは大きくテーリングしており,パラキシレンとメタキシレンの分離は困難であった。メタキシレンの分子径はMFIの細孔径よりもわずかに大きいことから,メタキシレンの極めて速い溶出と大きなピークテーリングは,少量のメタキシレンがMFI細孔内を極めて遅い速度で拡散している,もしくは,結晶表面(もしくは細孔開口部)に強く吸着していることを示唆している。これに対し,silicalite/ZSM-5およびsilicalite-1では,パラキシレンをメタキシレンから明確に分離することが可能であった。また,silicalite/ZSM-5のパラキシレンの保持時間はsilicalite-1よりもかなり大きい。保持時間の差はその物質の吸着の強さに依存することから,silicalite-1におけるパラキシレンの早い溶出は骨格内Alが存在していないことに起因する。silicalite/ZSM-5, silicalite-1いずれのサンプルにおいてもメタキシレンのピークテーリングはZSM-5と比較して極めて小さい。これらのサンプルの結晶外表面にはAlサイトが存在しておらず,より均一な表面を有していると考えられ,これがメタキシレンのピークテーリングの抑制に繋がったと言える。さらに,ゼオライト骨格中のAl含有量が増大すると,格子間隔が増大する23)。つまり,ゼオライトの細孔径はSi/Alが大きくなるにつれて,極めてわずかではあるものの小さくなるため,silicalite/ZSM-5の結晶外表面の細孔径はZSM-5と比較してわずかに小さく,これが分子の拡散に影響している可能性がある。実際,Fig. 4に示すように単成分蒸気吸着測定により,コアシェル構造化はパラキシレンの吸着速度に影響しない一方で,メタキシレンの吸着速度が大きく低下する。
次に,パルスクロマトグラフィー24)を用いてキシレン異性体のヘンリー定数および低被覆率時における分離係数(ヘンリー定数の比)を求めたところ(Table 2),コアシェル構造化により分離係数は3倍程度向上することがわかった。キシレン異性体それぞれのヘンリー定数から明らかなように,コアシェル構造化は細孔径よりも分子径の小さなパラキシレンへの影響は小さく,より分子径の大きいメタおよびオルトキシレンに対する吸着阻害効果がより強く現れる結果となった。なお,クロマトグラフィー分離で良好な分離性能を示したsilicalite-1の分離係数は最も低い値であった。これは骨格中にAlサイトが存在しておらず,パラキシレンのヘンリー定数が他のサンプルに比べ低いことに起因する。この結果は,高いパラキシレン分離係数の達成には,Alサイトのような骨格中の強吸着サイトの存在と,結晶外表面における強吸着サイトの不在が重要な鍵となることを示唆するものである。
Table 2. Henry adsorption constants of xylenes and low coverage separation factors on ZSM-5 and silicalite/ZSM-5 at 553 K | ZSM-5 | Silicalite/SM-5 | Silicalite-1 |
|---|
| K’(mol/kg/Pa) | Separation
factor* | K’(mol/kg/Pa) | Separation factor* | K’(mol/kg/Pa) | Separation factor* |
|---|
| p-xylene | 2.57×10−4 | | 1.66×10−4 | | 4.19×10−6 | |
| m-xylene | 4.46×10−5 | 5.8 | 9.94×10−6 | 16.7 | 9.50×10−7 | 4.4 |
| o-xylene | 3.48×10−5 | 7.4 | 7.31×10−6 | 22.7 | 1.04×10−6 | 4.0 |
| *determined by the ratio of Henry adsorption constants of p-xylene over other isomers |
2.3 表面疎水化による水蒸気共存下でのCO2分離25)
圧力スイングCO2分離プロセスではその高いCO2選択性からしばしば低シリカゼオライトが用いられる。しかしながら,低シリカゼオライトは水分子との親和性が高く,水蒸気共存下ではCO2よりも水分子が低シリカゼオライトに強吸着するため,CO2吸着量が大きく低下することが知られている。コアシェル構造化ゼオライトは,その結晶外表面が疎水的な純シリカゼオライトで構成されており,水蒸気共存下でのCO2分離への応用が期待できる。前節で用いたZSM-5をcoreとし,フッ化物法でコアシェル構造化したsilicalite (F)/ZSM-5およびフッ化物法を用いずに合成したsilicalite/ZSM-5について水蒸気共存下におけるCO2吸着性能を評価した。
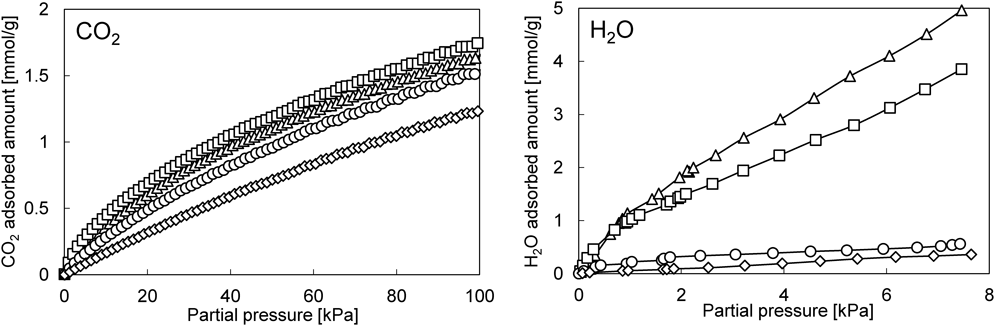
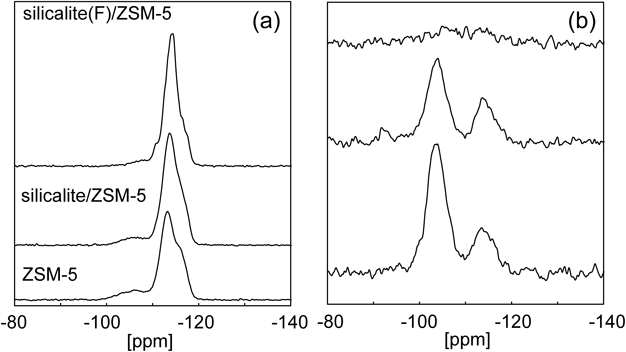
Fig. 5にそれぞれのサンプルおよび参照用としてフッ化物ありで合成したsilicalite-1(F)のCO2および水蒸気の単成分吸着等温線を示す。Silicalite/ZSM-5およびsilicalite (F)/ZSM-5のCO2吸着量はコアシェル構造化により元のZSM-5から多少の変化が認められるものの,その影響は小さく,coreであるZSM-5がAlサイトを有しているため,silicalite-1(F)よりも高い値を示した。一方で,水蒸気吸着に対するコアシェル構造化の影響はFig. 5に示すように非常に大きい。特にフッ化物法により合成したsilicalite (F)/ZSM-5の水蒸気吸着量は0.56 mmol/gとZSM-5の4.96 mmol/gから大幅に低下しており,silicalite-1 (F)と遜色ない低い水蒸気吸着量を示している。Silicalite/ZSM-5とsilicalite (F)/ZSM-5の水蒸気吸着量の大きな差は,silicalite-1層の骨格内欠陥によって説明できる。ゼオライト骨格内の格子欠陥はフッ化物を用いて合成することで大きく減少することが知られている26)。事実,Fig. 6に示すように,各サンプルの29Si MASおよびCP NMRを確認すると,silicalite/ZSM-5ではQ3(0Al)の存在が確認でき,一方のsilicalite (F)/ZSM-5では,29Si MAS NMRのQ3(0Al)ピークは減少しているのに加え,Q4(0Al)のピークがわずかに割れているのが確認できた。これはsilicalite (F)/ZSM-5の結晶性が他のサンプルよりも優れていることを示唆している。
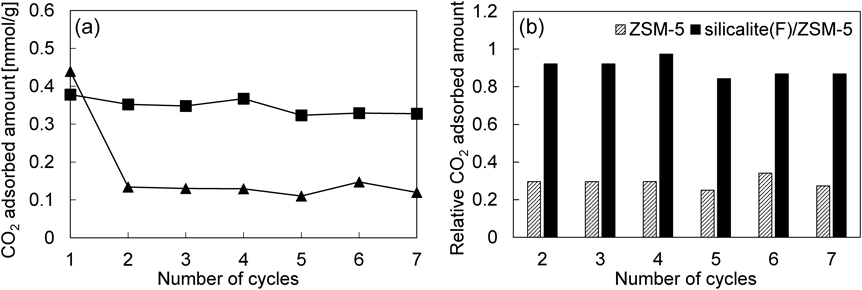
それぞれのサンプルを用い,水蒸気共存下におけるCO2吸着量を評価した。Fig. 7に吸脱着繰り返しによるCO2吸着量の変化を示す。ZSM-5は初期の吸着量は0.44 mmol/gであったのに対し,2回目の吸着量は0.13 mmol/gと大きく低下した。この大幅なCO2吸着量の低下は水蒸気の吸着によるものであり,熱再生によりCO2吸着量は初期と同程度まで回復する。これに対し,silicalite (F)/ZSM-5では,初期吸着量を基準とした2回目のCO2吸着量の低下は8%以下であり,7回目においてもその吸着量の低下はわずか13%であった。ZSM-5では初期吸着量の70%以上の吸着量が1回の吸脱着プロセスで失われていることを考慮すると,silicalite (F)/ZSM-5は優れたCO2選択性を有していると言える。
3. CHA型ゼオライトのコアシェル構造化による吸着選択性の向上27)
MFI型ゼオライトのコアシェル構造化は著者らだけでなく多くの研究報告があり,比較的合成も容易である。ゼオライトのコアシェル構造化は他のゼオライトへの応用も原理的には可能であるが,その報告例は未だなされていない。筆者らは本手法の汎用性を実証するため,CHA型ゼオライトのコアシェル構造化を試み,ブタノール発酵の主生成物であるアセトン–ブタノール–エタノール(ABE)系の分離可能性を評価した。
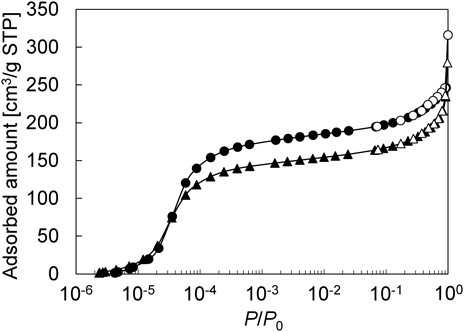
CHA型ゼオライトのコアシェル構造化は前述のMFI型と同様にフッ化物を用いて合成が可能である。Fig. 8にcoreとして用いたCHA型アルミノシリケート(parent CHA)とコアシェル構造化CHA型ゼオライト(coated CHA)のSEM像を示す。いずれのサンプルも擬立方晶形状を有していた。SEM像から見積もった平均結晶径は3.87 µmおよび4.06 µmとコアシェル構造化によりわずかに結晶径は大きくなり,Si/Alも39.3から40.6へ増大していた。Fig. 9に示すようにいずれのサンプルもAr吸着等温線はI型を示していたが,parent CHAおよびcoated CHAのミクロ孔容積はそれぞれ0.21 cm3および0.25 cm3とparent CHAのミクロ孔容積は低い値を示していた。この原因は恐らくサンプル中に存在しているアモルファス様物質によるものと考えている。次に,パラキシレンのクラッキング(400°C)により結晶外表面の触媒活性を評価したところ,コアシェル構造化によりパラキシレン転化率は41%から0.2%まで低下することを確認した。これらの結果は,CHA型アルミノシリケートの結晶外表面を純シリカCHA薄層で被覆できていることを示唆している。
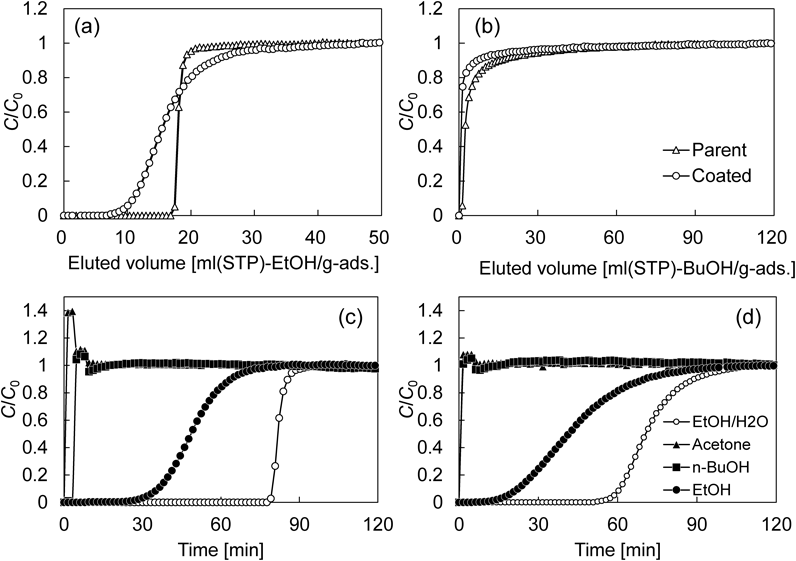
Parent CHAおよびcoated CHAを用いて,アルコール/水蒸気二成分系の動的吸着特性を評価した。Fig. 10(a)に示すように,Parent CHAでは破過曲線の鋭い立ち上がりが確認されたのに対し,uncoated CHAの破過曲線の立ち上がりは非常に緩やかであった。これは純シリカCHA被覆層がエタノールの吸着抵抗になっていることを意味している。1-ブタノールの場合,分子サイズが細孔径と同程度であることから,立体障害により破過時間は非常に短く,破過曲線の立ち上がりも鋭い結果となっている(Fig. 10(b))。加えて,コアシェル構造化により破過がより早くなっていることから,コアシェル構造化が1-ブタノールに対する分子ふるい効果を促進していることが示唆される。
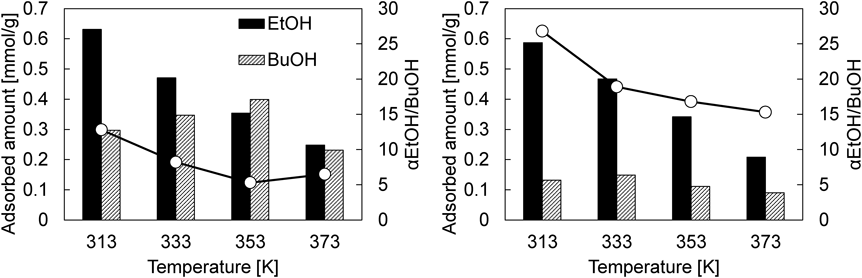
次に,ABE/水蒸気多成分系で評価したところ,parent CHAにおけるアセトンの破過は極めて早い。これはアセトンの分子径がCHAの細孔径に比べ十分に大きいことに起因する28)。エタノールに注目すると,エタノール/水蒸気二成分系と比較して,その破過曲線はより緩やかな立ち上がりを示した。これは,わずかに吸着する1-ブタノールの細孔内拡散は極めて遅く29),吸着した1-ブタノールがエタノールの細孔内拡散を抑制していることに起因する。Coated CHAに注目すると,上述のparent CHAの場合と同様の傾向が見られるのに加え,二成分系の場合と同様,1-ブタノールの破過はコアシェル構造化により早くなり,1-ブタノールはアセトンと同時に破過していた。この結果は,多成分系においてもコアシェル構造化によって,1-ブタノールの吸着が阻害されていることを意味している。4成分系におけるエタノールおよび1-ブタノールの吸着量と分離係数をFig. 11にまとめた。エタノール吸着量はコアシェル構造化によりわずかに低下するものの,ほぼ同程度であった。これに対し,1-ブタノールの吸着量はコアシェル構造化により0.28 mmol/gから0.12 mmol/g(40°C)へと減少し,その結果,エタノール/1-ブタノール分離係数は12.8から26.8へと向上することがわかった。さらに,parent CHAの場合,1-ブタノール吸着量に対する温度の影響は顕著に確認され,40°Cから80°Cで1-ブタノール吸着量は増大し,その後,減少する傾向を示した。温度上昇による吸着量の増大は,1-ブタノールの細孔内拡散が改善したことによるものと考えられ,その後,100°Cにおける吸着量の減少は,温度上昇による平衡吸着量の低下に起因しているものと考えている。一方で,coated CHAでは1-ブタノール吸着量に対する温度の影響はほとんど認められず,いずれの吸着温度でもその吸着量は低いままであった。これらの結果は,純シリカCHA層が明らかに拡散抵抗となり,特に1-ブタノールに対する吸着阻害効果を示すものである。
本稿では,ゼオライトのコアシェル構造化が吸着特性に及ぼす影響について紹介した。低シリカゼオライトの外表面への純シリカゼオライトの被覆は,①結晶外表面の強吸着サイトの不在もしくは細孔開口部がわずかに狭小化することによる分子ふるい能の促進や,②結晶外表面の疎水化による水蒸気吸着阻害効果が得られることを明らかにした。また,本研究ではじめてCHA型ゼオライトのコアシェル構造化を試み,MFI型ゼオライトの場合と同様の分子ふるい促進効果が得られることを示し,ゼオライトのコアシェル構造化が吸着性能向上の汎用的な手法となり得ることを実証した。
謝辞Acknowledgments
本研究は,本学共同研究者およびVrije Universiteit BrusselのProf. Joeri F.M. Denayerとの共同研究によって行われたものです。また,科学研究費助成制度(26820337, 15KK0234)の支援を受けて実施されました。
引用文献References
1) Y. Tao, H. Kanoh, L. Abrams, K. Kaneko, Chem. Rev., 106, 896(2006).
2) M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki, R. Ryoo, Nature, 461, 246(2009).
3) T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo, T. Tatsumi, J. Phys. Chem. C, 119, 15303(2015).
4) Z. Lai, G. Bonilla, I. Diaz, J. G. Nery, K. Sujaoti, M. A. Amat, E. Kokkoli, Science, 300, 456(2003).
5) P. Huttenloch, K. E. Roehl, K. Czurda, Environ. Sci. Technol., 35, 4260(2001).
6) P, Loganathan, S. Vigneswaran, J. Kandasamy, J. Environ. Manage., 131, 363(2013).
7) N. M. Mahmoodi, M. H. Saffar-Dastgerdi, Microchem. J., 145, 74(2019).
8) S. J. Reitmeier, O. C. Gobin, A. Jentys, J. A. Lercher, Angew. Chem. Int. Ed., 48, 533(2009).
9) W. W. Kaeding, C. Chu, L. B. Young, B. Weinstein, S. Butter, J. Catal., 67, 159(1981).
10) N. Y. Chen, W. W. Kaeding, F. G. Dwyer, J. Am. Chem. Soc., 101, 6783(1979).
11) M. Niwa, M. Kato, T. Hattori, Y. Murakami, J. Phys. Chem., 90, 6233(1986).
12) A. B. Halgeri, J. Das, Catal. Today, 73, 65(2002).
13) J. Čejka, N. Zilková, B. Wichterlová, G. Eder-Mirth, J. A. Lercher, Zeolites, 17, 265(1996).
14) S. Inagaki, K. Sato, S. Hayashi, J. Tatami, Y. Kubota, T. Wakihara, ACS Appl. Mater. Interfaces, 7, 4488(2015).
15) M. Miyamoto, T. Kamei, N. Nishiyama, Y. Egashira, K. Ueyama, Adv. Mater., 17, 1985(2005).
16) Y.-Q. Deng, W.-F. Zhou, H.-M. Lv, Y.-Y. Zhang, C.-T. Au, S.-F. Yin, RSC Adv., 4, 37296(2014).
17) A. Ghorbanpour, A. Gumidyala, L. C. Grabow, S. P. Crossley, J. D. Rimer, ACS Nano, 9, 4006(2015).
18) M. Li, Y. Hu, Y. Fang, T. Tan, Catal. Today, in press(2019)DOI: 10.1016/j.cattod.2019.02.041.
19) D. V. Vu, M. Miyamoto, N. Nishiyama, S. Ichikawa, Y. Egashira, K. Ueyama, Microporous Mesoporous Mater., 115, 106(2008).
20) M. Miyamoto, S. Ono, Y. Oumi, S. Uemiya, S. Van der Perre, T. Virdis, G. V. Baron, J. F. M. Denayer, ACS Appl. Nano Mater., 2, 2642(2019).
21) M. Okamoto, Y. Osafune, Microporous Mesoporous Mater., 143, 413(2011).
22) K. Nakai, J. Sonoda, M. Yoshida, M. Hakuman, H. Naono, Adsorption, 13, 351(2007).
23) J.-H. Kim, T. Kunieda, M. Niwa, J. Catal., 173, 433(1998).
24) L. I. Devriese, J. A. Martens, J. W. Thybaut, G. B. Marin, G. V. Baron, J. F. M. Denayer, Microporous Mesoporous Mater., 116, 607(2008).
25) M. Miyamoto, S. Ono, K. Kusukami, Y. Oumi, S. Uemiya, ChemSusChem, 11, 1756(2018).
26) S. A. Axon, J. Klinowski, Appl. Catal. A, 81, 27(1992).
27) M. Miyamoto, H. Iwatsuka, Y. Oumi, S. Uemiya, S. Van den Perre, G. V. Baron, J. F. M. Denayer, Chem. Eng. J., 363, 292(2019).
28) S. Van der Perre, P. Gelin, B. Claessens, A. Martin-Calvo, J. Cousin Saint Remi, T. Duerinck, G. V. Baron, M. Palomino, L. Y. Sánchez, S. Valencia, J. Shang, R. Singh, P. A. Webley, F. Rey, J. F. M. Denayer, ChemSusChem, 10, 2968(2017).
29) J. Cousin Saint Remi, G. V. Baron, J. F. M. Denayer, J. Phys. Chem. C, 117, 9758(2013).