International Zeolite Association(IZA)は,2019年4月現在で248のゼオライト骨格を承認している1)。ゼオライトの合成の歴史と最先端の技術はいくつかの総説にまとめられている2–7)ものの,デザインされた合成となるとまだまだ達成されているとは言い難い。ゼオライト合成に関する近年の動向は,次のように大まかに分類することができる4):(i)新規なorganic structure-directing agent(OSDA)の開発5),(ii)OSDAを用いない(=OSDA-free条件での)合成8,9),(iii)超疎水性物質の合成10),(iv)層状前駆体(二次元物質)から三次元物質への転換またはその逆,例えばassembly-disassembly-organization-reassembly(ADOR)と呼ばれる手法11),(v)階層構造ゼオライト12),(vi)キラル材料5,13–16),および(vii)特定のT-siteへのヘテロ原子の配置17–19)。小細孔ゼオライトの重要性も近年増大している20)。上記の例のうち特にゲルマノシリケートは,骨格中のGe-O結合は通常,加水分解に弱く安定性に欠ける。一方でGe-O結合の不安定性を逆手にとって材料を新しい構造に変換するために利用する合成戦略もある21,22)。とは言え,ゼオライトのイオン交換剤や固体酸としての応用を考慮すると,やはりアルミノシリケートが実用上好ましい。ところが,2000~2017年に承認された106個の新しいゼオライト骨格のうち,実用的に有用な多次元,大細孔,アルミノシリケート系骨格はごく少数しかない。具体的にはMCM-6823,24)(MSEトポロジー,12–10–10-ring), ITQ-2725)(IWVトポロジー,12–12-ring), SSZ-6526)(SSFトポロジー,12–12-ring)だけである。
このような状況に鑑みて,我々は,工業化されていないが非常に有望なアルミノシリケートであるMCM-68に注目し,触媒適用に向けた合成研究を始めた。このことは以前に触媒誌でも紹介した27)。MCM-68が検討に値する理由は以下のとおりである。1980年代以降,Zonesらは複雑な構造を持つ様々なOSDAを合成し,水熱合成法による多くの新しいゼオライトの合成を実現した。2000年以前に報告された高シリカゼオライトの大部分は,より低いdimensionalityを有している28)ために一般に拡散性に関して劣っており,したがって触媒材料としての用途が制限されていた。さらに,生成物はBをAlで置き換える手順を必要とするボロシリケートである場合が多く,Alの導入量は十分でなかった29,30)。ナノ粒子に準じる小さい粒径も触媒用途に有利な要因である。MSE骨格を有するMCM-68は,これらの要請をほとんど満たし,期待どおりに種々の触媒用途に対して有望な結果をもたらしてきた31,32)。例えば,適度に脱アルミニウム(脱Al)されたMCM-68はヘキサンの接触分解33)およびdimethyl ether(DME)-to-olefin(DTO)反応のための触媒として特に有用であることがわかった34)。このように,MSE型アルミノシリケートに関する一連の触媒研究によって,MCM-68および関連材料が固体酸触媒としていかに有用かがわかってきた。
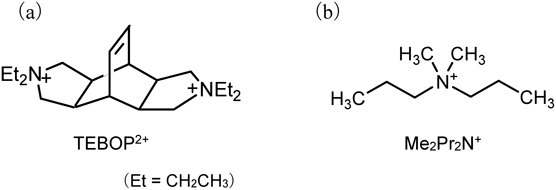
この時点で,次に挙げる課題が残った:(1)結晶化時間の短縮,(2)従来型OSDAを単純化することによるN,N,N′,N′-tetraethylbicyclo[2.2.2]octa-7-ene-2,3:5,6-dipyrrolidinium(TEBOP2+)(図1a)からの脱却。このうち(1)は,FAU型ゼオライトのいわゆる「水熱転換法」によってMSE骨格の結晶化を促進することが予想された。検討の結果,それまで達成されていなかったAlリッチな化学組成(Si/Al=6.9)を有するMSE型ゼオライトが,従来の約1/3の時間(典型的には5日)で結晶化した35)。FAUとTEBOP2+を組み合わせた条件でAFXの結晶化も達成され,高性能AFX触媒の創製に成功した36)。この結果は,水熱転換法を用いると従来の水熱合成では達成できなかったことを実現できることを示していたため,さらなる展開を図った。
一方,Universal Oil Products(UOP)の研究者らは,より単純なOSDAとしてdimethyldipropylammonium(Me2Pr2N+)(図1b)を用いたUZM-35ゼオライト(Si/Al=8, MSEトポロジー)の開発を行っていた37)。MSE骨格の「Type Material」であるMCM-68と比較して,UZM-35の合成に用いるOSDAの単純さが魅力的であった。ただし,単純なOSDAの宿命であるが,UOPが開示している手順ではUZM-35の結晶化の際に多くの競合相が出現した。そこで我々は,出発物質としてFAU型ゼオライトを使用する水熱変換法を適用した。その結果,LTL,ANAおよび正長石(orthoclase)などの不純な相の形成がうまく回避され,MSEの速い結晶化が実現できた。
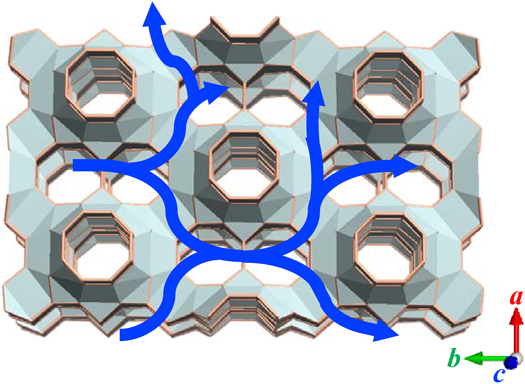
ここで我々は,濃厚系から出発するゼオライト合成に興味を持った。従来の水熱合成と,乾燥ゲルを使用するdry-gel conversion(DGC)法38)との間の未開拓領域である。その結果,Me2Pr2N+を用いて,YNU-539)の合成に成功した。YNU-5は我々が世界で初めて合成した新しい大細孔アルミノシリケートゼオライトであり,12–12–8員環細孔系と孤立した8員環ストレートチャンネルを含む独特の細孔系を有している(図2)。この新しい骨格に対しては,IZAによってYFIという骨格コード1)が承認された。YNU-5の合成のための詳細な骨格構造および詳細な合成手順がすでに報告されている39,40)。また,合成後処理による骨格の熱安定性の向上について興味深い挙動が見られているため,本稿ではそれを重点的に解説したい。なお,ゼオライトの員環数はIZAが推奨するとおり,例えば酸素8員環の場合8-ringと表記することとする。日本語の場合,本稿では敢えて「酸素」を省略し,「8員環」と表記する。また,YNU-5 (57)のようにゼオライト名の直後に括弧付きで数値を示した場合,その数値「57」はSi/Al比を意味している。
2.1 水の仕込み量がstructure-directionに及ぼす影響
我々は以前,比較的小さなOSDAと濃厚な合成液から出発する手法とを組み合わせて,約16のSi/Al比を有するSSZ-39(AEI)の合成に成功した41)。AEIとしては最大レベルのSi/Alである。濃厚溶液を使用する戦略を採用したのは,比較的小さく親水性のOSDAの可能性を引き出すのに効果的ではないかとの期待からである。その際に上述のようにMe2Pr2N+というフレキシブルで疎水性の低いOSDAを用いたわけである。当初,痕跡量の不純相(後にYNU-5と判明)が存在していたが,この時点で同定には至らなかった。仕込みと生成相の変化を注意深く検討した結果,徐々にこのunknown phaseの生成比が増大し,主生成物として得られるようになった。ここが合成を成功に導いたもっとも重要なポイントである。吸着挙動や化学分析の結果などからマイクロポーラスなアルミノシリケートであることが確かめられた一方で,XRDパターンは依然として既知のゼオライト相には帰属できなかった。このことから,この未知のゼオライト相が新規骨格を有するのではないかという可能性が膨らんだ。そこで,ほぼ純粋となったサンプルを用いて,産総研・池田博士による構造決定がなされ,YNU-5の構造が明らかとなった39)。大まかな手順としては,焼成試料について指数付けによって空間群と格子定数を得た後,最大エントロピーパターソン法,直接法,powder charge-flipping法を併用し,骨格トポロジー情報を得た。トポロジーの確からしさは高分解能TEM/STEM観察によっても確認された。構造解析の詳しい経緯については,別の解説に譲りたい。
YNU-5の合成において最も顕著だったのは出発ゲル中の水の量の影響である。合成混合物中のH2O/SiO2比(ここではxと置く)が生成物相に及ぼす影響を図3に示す。出発ゲル組成は0.265(SiO2)FAU−0.735(SiO2)Ludox−0.025(Al2O3)FAU−0.17 Me2Pr2N+OH− −0.15 NaOH−0.15 KOH−x H2Oであり,(SiO2)FAUと(SiO2)LudoxはそれぞれFAU型ゼオライトおよびコロイダルシリカ(Ludox AS40)由来のSiO2である。比較的水リッチなx=10のとき,MFI相が結晶化した。水の量をわずかに減らしていくと,x=7付近の狭い範囲で純粋なYNU-5が結晶化した。次いでx=4まで水を減らすと今度はMSE相が結晶化した。Me2Pr2N+を用いたMSE型ゼオライトの生成は,生成物がUZM-35として知られているのでこれは不思議ではない35)(ただし,我々の場合は実際にはUZM-35を得るために出発物質としてFAU型ゼオライトを使用して独自の合成手順を考案した)。水をさらに減らしてx=3とすると,*BEA相が現れた。我々の知る限りでは,OSDAとしてMe2Pr2N+のみを用いてベータゼオライトが得られた最初の事例であり,驚くべき結果である。同じ分子量およびC/N+モル比を有するテトラエチルアンモニウムは,非常に広い合成範囲で*BEA相を結晶化することが知られているのに対し,Me2Pr2N+を使用すると*BEA相は結晶化しないものと認識されてきたからである。全体として,生成相の選択に対してxの値がこれほどまでに影響することは従来の常識では考えられず,予想外であった。なお,ここに示した一連の検討ではMOR相が現れなかったが,MOR相はむしろ出現しやすい競合相であることを付記する。例えば,原料FAUのロット変更に伴う微妙な条件変化によりMOR相が現れやすくなる。
仕込みH2O/SiO2比(x)と骨格密度(framework density; FD)との間には良好な相関関係が見出された。FDの代表値としては,FDsi値(T/nm3)1)を使用した。MFI, MSEおよび*BEA相に対する(FDsi, x)値は,それぞれ(18.4, 10),(16.4, 4)および(15.3, 3)である。したがって,主生成物相のFDは,合成混合物中の水の量が減少するにつれて減少する。出発ゲルのxが大きいほど大きいFDを有するゼオライト相の結晶化を促進し,これはおそらくOSDAとケイ酸塩との間の相互作用の度合いを反映する。濃厚な合成液では,ケイ酸塩種とOSDAはより効率的に相互作用するので,より多くのOSDAが組み込まれる。別の観点からは,疎水性相互作用を強めるために濃厚系は有利であり,これが功を奏して新しい大細孔ゼオライトYNU-5が単純なOSDAの使用により結晶化する領域が見出されたとも言える。無機および有機種の両方によるstructure-directionは,YNU-5の結晶化に必須であると思われる。有機種は孤立した8員環ストレートチャンネルに入ることができないため,YNU-5の孤立した8員環ストレートチャンネルはK+で満たされている39)。OSDAは12員環のチャンネルに包接されており,おそらく孤立8員環を含むビルディングブロックを集めて配列させる役割を果たしていると考えられる39)。
2.2 結晶化の経時変化
静置条件下でのYNU-5の結晶化過程におけるXRDの経時変化を図4に示す。16時間後には出発物質として用いたFAUのパターンだけが見られ,同時にアモルファスハローがわずかに観察された。バルクSi/Al比は7.2で,仕込み比よりわずかに増加した。この時点で,Na/AlおよびK/Al比はそれぞれ0.25および0.39であり,K+がすでに優先的に取り込まれていることを示している。28時間後,アモルファス相は消失し,FAUとYNU-5の混合物が得られた。これはYNU-5の結晶化がFAU共存下で進行したことを示唆している。45時間後,YNU-5の結晶化は完了し,その時点でSi/Al, Na/AlおよびK/Al比はそれぞれ9.5, 0.11および0.55であった。結晶化時間を165時間に延ばしても,YNU-5は単相のままであった。
2.3 酸処理によるSi/Al比の制御
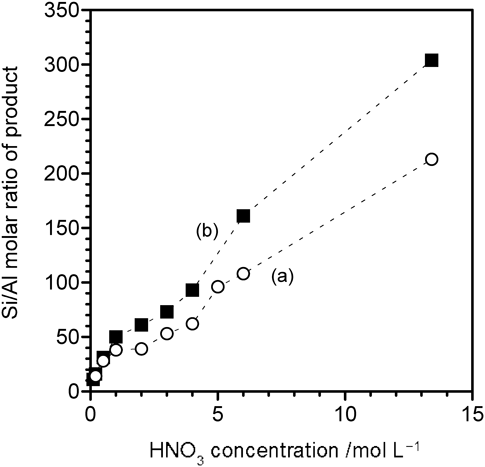
種々の濃度(0.1~13.4 mol/L)の硝酸処理による脱Al後も骨格構造は維持された。また,これらの処理によりアルカリカチオンはプロトンに交換された。図5は,酸処理濃度とSi/Al比の相関図である。図5aは80°C,2 hの条件【条件A】,図5bは130°Cの油浴で24 h加熱する条件【条件B】(我々はこれを便宜上 reflux, 24 hと表現している)で脱Alした結果を示す。これらの結果は,単に酸濃度を増加させることで,Si/Al比も30から>300までの範囲で単調増加することを示している。単純な酸処理による脱Alは場合によっては困難であるが,YNU-5は幸い*BEAおよびMSEのような他のペンタシル型ゼオライトと同様に,容易に脱Alされることがわかった。同じ酸濃度で比べた場合,【条件B】の方が【条件A】よりも脱Al度は当然高くなり,Si/Al比300以上の高度な脱Alは【条件B】でのみ可能であった。
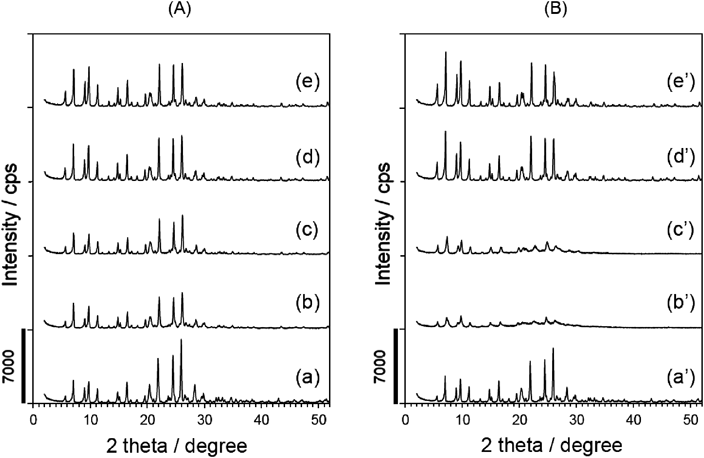
ゼオライトの脱Alは欠陥(欠損)部位(site defect)を形成するため,骨格安定性低下が懸念される。そこで,脱Al試料の熱安定性を調べた。脱Al試料を650°Cで熱処理した前後の粉末XRDパターンを図6に示す。脱Al処理されていないサンプルは十分な熱安定性を示した(図6a,6a’)。80°Cで処理したサンプル(図6b,6cに相当)では,熱処理後のXRD(図6b’,6c’)でわかるように結晶化度がかなり低下し,これは脱Alによって生じた欠損の影響と考えられる。興味深いことに,【条件B】で処理したサンプル(図6d)は,高い熱安定性を示した(図6d’)。これらの結果は以下のようにまとめることができる。80°C【条件A】で脱Al処理した試料の結晶化度は熱処理後にかなり低下するのに対し,>100°Cで脱Alした試料は高い熱安定性を有し,650°Cでの熱処理後に結晶性が全く失われない。Si-migration42,43)が骨格の安定化に何らかの役割を果たしていると考えられる。
3.1 固体NMRによる検討
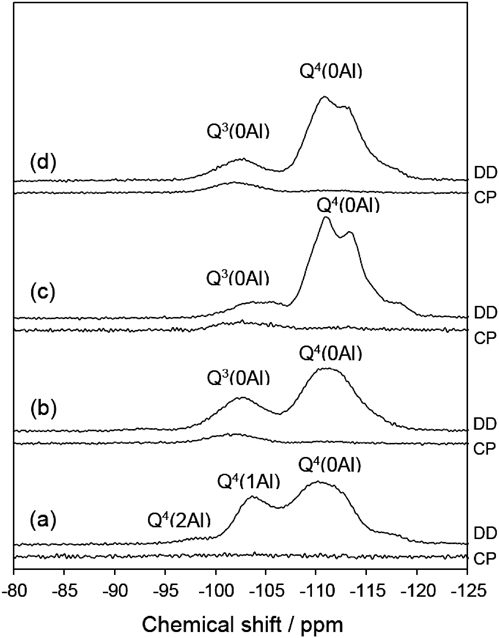
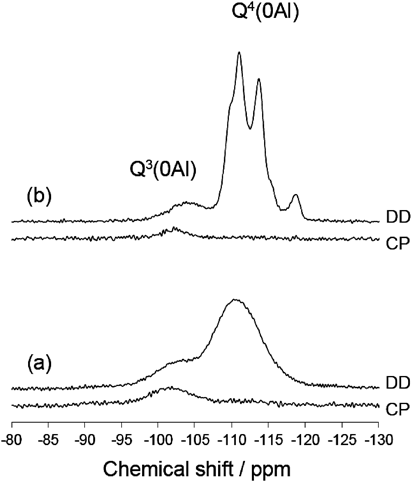
29Si MAS NMR(図7)によって欠損の量を推定した。ここでSi(OSi)x-y(OAl)y(OH)4-xをQx(yAl)と略す44)。酸処理前には,Q3(0Al)ピーク,すなわちSi(OSi)3(OH)ピークは観察されなかった。より穏やかな条件下(80°C,2 h;条件A)で処理したサンプルのスペクトルでは,大きなQ3(0Al)ピークが存在した。Q3/Q4ピーク面積比は31/69であった。一方,より苛酷な条件下(reflux,24 h;条件B)で処理したサンプルでは,Q3(0Al)ピークがはるかに小さかった。Q3/Q4比は19/81であり,より厳しい条件下で欠損が減少したことを示している。
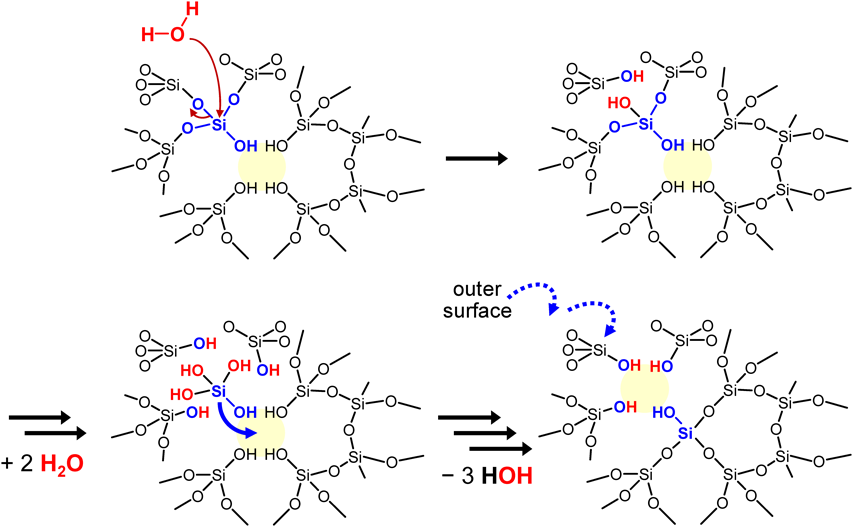
これらの結果は,我々が骨格安定化の原因として考えているSi-migrationで説明できる42,43)。我々はこれまで,500°Cを超える水蒸気処理による骨格安定化を見てきたが,今回の液相条件は強い方の【条件B】であっても,水蒸気処理に比べて圧倒的に穏やかであり,この液相条件下でのSi-migrationによる骨格安定化は予想外であった。欠損数の減少のメカニズムは以下のように説明できる。最初,欠損に隣接するSi-Oが酸加水分解され,生じたモノケイ酸種が隣の欠損部位(=シラノールネスト)に移動して再び縮合する。これは,図8に示すように,隣接サイトへの欠損の見かけ上の移動に対応する。このプロセスを何度も繰り返した後,欠陥サイトは最終的に結晶の外面に到達する。同様の安定化プロセスがスチーム処理プロセスにおいて想定されていた45)。今回の我々の結果は,酸水溶液中で100°Cというスチーミングに比べて圧倒的に低温の酸処理中にもSi-migrationが起こることを示している。80°Cまで下げるとSi-migrationが遅くなるが,脱Alはこの低温でも十分に速い。この推測は,SkeelsおよびBreckが(NH4)2SiF6処理の場合に比較的速い脱Alおよび欠損へのより遅いSi導入を報告している46)ことと合致する。100°Cに昇温することでSi-migrationはより速くなり,すでに速い脱Alによって生じた欠損をすばやく修復することができたものと推測できる。
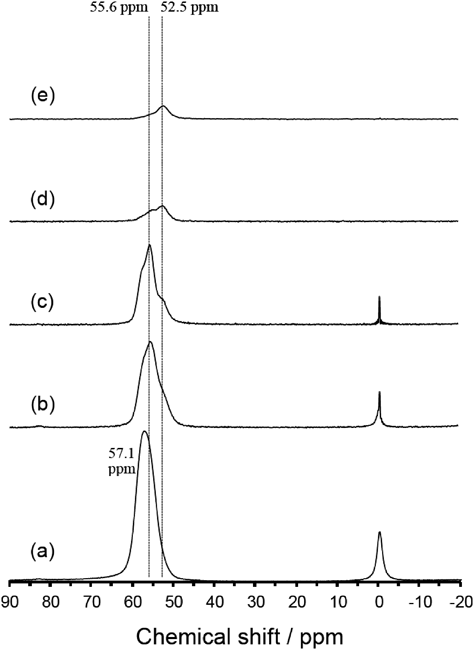
27Al MAS NMR(図9a~9c)には,四配位Alに帰属できる50~60 ppmの主ピークがはっきりと見られる。0 ppm付近には骨格外Al(6配位Al)に起因する小さなシグナルがあるが,ここでは50~60 ppm付近のピークについてのみ説明する。高度に脱AlしたYNU-5以外のスペクトルでは,主ピークが約56 ppmに存在し,約53 ppmに肩が見られる。高度脱Al体のスペクトルでは,相対強度が逆転し,53 ppm付近のピークが単一となった。これは,孤立した8員環ストレートチャンネル内の骨格Al原子を酸加水分解によって除去するのが相対的に困難であり,最後まで残る骨格Alが53 ppm付近のピークに相当することを示唆している。孤立した8員環ストレートチャンネルからの脱Alが比較的困難であるのは,そのチャンネルへの水分子の拡散が著しく制限されているためと考えられる。文献47によれば,化学シフトはフレームワーク中のT–O–T角(T=Al or Si)の関数であり,27Al MAS NMRスペクトル中の高磁場(右側)ピークは以下のように,より大きいT–O–T角に帰属できる。孤立した8員環ストレートチャンネルの内側にのみ位置するT1とT5の場合,結晶学的データに基づくと,T–O–T角は他のTサイトよりも大きくなる39)(Tサイトの番号付けも文献39のとおり)。T–O–T角に基づくこの帰属は,孤立した8員環ストレートチャンネルよりも12員環チャンネルに対して水分子(オキソニウムイオン)は接近しやすいであろうという推論に基づく帰属と矛盾しない。
3.2 窒素吸着挙動
焼成体,H+交換体,および高度に脱AlしたYNU-5サンプルは,IUPACによる典型的なI型の窒素吸脱着等温線を示し,比表面積やt-plot法によって見積られるミクロ細孔容積の有意な変化はなかった。高度脱Al体(=安定化サンプル)では,メソ孔形成の兆候はほとんど見られなかった。700°Cでの水蒸気処理によるOSDA-free *BEAおよびOSDA-free MSEの安定化に関する以前の検討では,わずかなメソ孔形成が見られ(文献43の図S2およびS13参照),さらに厳しい条件下では顕著なメソ孔形成が観察された(文献32の図S4参照)。これらと比較し,今回得られた結果は,100~130°C程度の低い温度での液相処理であり,ずっと穏やかな条件下であるので,メソ孔形成が目立たなくてもおかしくはない。
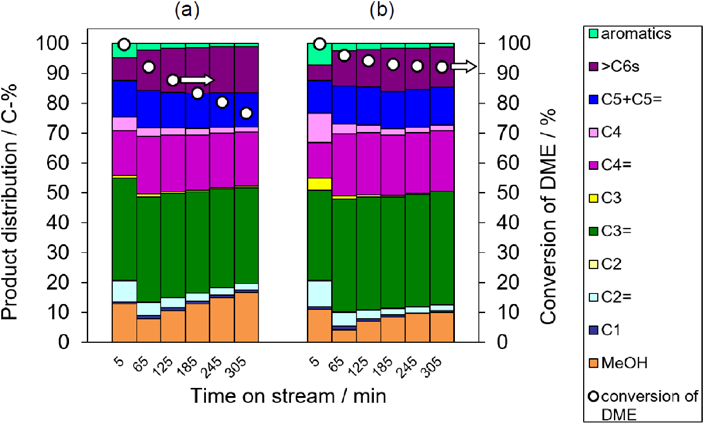
YNU-5の骨格安定化の効果を実証するために,dimethyl ether(DME)-to-olefin(DTO)反応に対する触媒性能を比較した。DTO反応は,非石油化石資源から低級オレフィンを得るための重要な反応である。触媒反応に用いる試料は,400 kg cm−2で加圧成形したものを500~600 µmに整粒して用いた。DTO反応は固定床流通式反応装置を用いて実施した。石英管に触媒100 mgを充填し,空気流通下で550°C, 1時間前処理を行った(この前処理により,NH4+-YNU-5はH+-YNU-5となる)。反応は400°Cで実施した。また,キャリアガスとしてHeを用い,W/F値は20 g-cat h mol−1とした。Time on stream(TOS)5分の時点から60分ごとにサンプリングした反応混合物をGC(FID)で分析した。結果を図10に示す。安定化処理前後のYNU-5触媒両者について,DMEの転化率はTOS 5 minの段階で十分に高かった。このうち安定化処理をしていない触媒の場合,TOSが増加するにつれて転化率が比較的急速に低下した(図10a)。ところがこの転化率の低下は,YNU-5(57)の骨格を安定化させることによって期待どおりにうまく抑制された。図10bの結果は,骨格が損なわれていないときの典型的な触媒挙動であり,それは以下のように説明することができる。YNU-5をSi/Al=57に脱Alすることによって,外表面または開口部(細孔内では優先的に酸素12員環)に存在する酸点を選択的に除去することができたが,細孔内の酸点は保持された。単独8員環の細孔入口の酸性部位は依然として残っており,コーク形成の原因となっている。図10bにおいてTOSが5から185分の間にあるとき,わずかな不活性化が観察され,これは単独8員環の細孔入口でプレコーキングが起こり,8員環の完全な不活性化後に12員環内に残った酸点によって触媒活性が保たれる。これに対して,安定化されていないYNU-5(57)を触媒とした場合,転化率の単調減少が観察された。この場合,反応後に回収された触媒の結晶性が著しく低下していることがわかった。
以上より,ゼオライト骨格を安定化するような条件でポスト処理をすることが今後の触媒開発に必須であり,有利であることがわかった。
5. 550°Cでの熱処理による骨格の規則性の向上
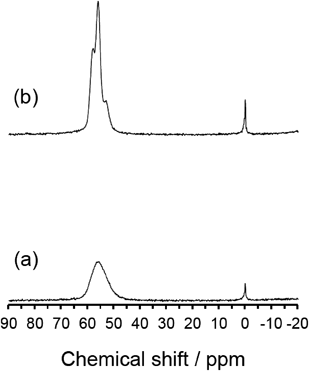
触媒反応を行う前の前処理温度である550°Cでの熱処理中のYNU-5の挙動に関して重要な結果が得られた。熱処理の前後で,粉末XRDパターンの変化は,図6で見てきた挙動と一致した。図11および図12にそれぞれ示すように,安定化YNU-5 (57)の29Siおよび27Al MAS NMRスペクトルに顕著な効果が観察された。すなわち,安定化されていないYNU-5 (60)ではブロードであるが,安定化されたYNU-5 (57)では550°Cで処理した後,ピークは著しく先鋭化した。これは,YFI骨格におけるユニットセルごとの原子位置のゆらぎが小さくなったためではないかと考えている。よく分離したピークは,Tサイトの区別,ひいてはヘテロ原子のサイト選択的な導入へ向けて,希望を与えるものと言える。
我々が最近合成に成功した新規骨格ゼオライトYNU-5の触媒応用へ向けた骨格安定化に焦点をあてて概説した。日本初のゼオライト新規骨格の種類は極めて少なく,欧米諸国に対して我が国は決定的な劣勢に立たされている。巻き返しの第一歩として,この分野への多くの若者たちの参画を期待したい。もちろん,単に新しい骨格というだけでなく,新たな機能の創生が望まれることは言うまでもない。概念的に斬新・画期的で効率の極めて良い合成手法が学術分野では待望される一方で,この30年間に米国の石油メジャーが行ってきたオーソドックスな手法による実用化事例の中に参考にすべきファクターがあるように思われる。本稿で紹介した成果は主としてJST戦略的創造研究推進事業チーム型研究(CREST)「超空間制御に基づく高度な特性を有する革新的機能素材等の創製」の支援のもとに得られたものである。ゼオライトの構造解析に関しては,池田拓史博士(産総研),日吉範人博士(産総研),合成と反応に関しては韓喬氏(横浜国大),吉田友香氏(横浜国大),淺沼開氏(横浜国大),その他大勢の方々のご協力を賜った。ここに記して謝意を表するとともに,記すことができなかった方々にも感謝したい。
引用文献References
1) Ch. Baerlocher, L. B. McCusker, D. H. Olson, Atlas of Zeolite Framework Types, 6th ed., Elsevier, Amsterdam(2007); see also: http://www.iza-structure.org/databases/.
2) M. E. Davis, R. F. Lobo, Chem. Mater., 4, 756(1992).
3) C. S. Cundy, P. A. Cox, Chem. Rev., 103, 663(2003).
4) M. E. Davis, Chem. Mater., 26, 239(2014).
5) J. Li, A. Corma, J. Yu, Chem. Rev. Soc., 44, 7112(2015).
6) Y. Li, J. Yu, Chem. Rev., 114, 7268(2014).
7) Z. Wang, J. Yu, R. Xu, Chem. Soc. Rev., 41, 1729(2018).
8) X. Meng, F. -S. Xiao, Chem. Rev., 114, 1521(2014).
9) K. Itabashi, Y. Kamimura, K. Iyoki, A. Shimojima, T. Okubo, J. Am. Chem. Soc., 134, 11542(2012).
10) Y. Sun, W. Lu, Y. Li, Appl. Phys. Lett., 105, 121609(2014)and references cited therein.
11) W. J. Roth, P. Nachtigall, R. E. Morris, J. Čejka, Chem. Rev., 114, 4807(2014).
12) D. P. Serrano, J. M. Escola, P. Pizarro, Chem. Soc. Rev., 42, 4004(2013).
13) A. Rojas, O. Arteaga, B. Kahr, M. A. Camblor, J. Am. Chem. Soc., 135, 11975(2013).
14) Y. Ma, P. Oleynikov, O. Terasaki, Nature Mater., 16, 755(2017).
15) S. K. Brand, J. E. Schmidt, M. W. Deem, F. Daeyaert, Y. Ma, O. Terasaki, M. Orazova, M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 114, 5101(2017).
16) M. E. Davis, ACS Catal., 8, 10082(2018)
17) A. Bhan, E. Iglesia, Acc. Chem. Res., 41, 559(2008).
18) J. Shin, D. S. Bhange, M. A. Camblor, Y. Lee, W. J. Kim, I. S. Nam, S. B. Hong, J. Am. Chem. Soc., 133, 10587(2011).
19) T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo, T. Tatsumi, J. Phys. Chem. C, 119, 15303(2015).
20) M. Dusselier, M. E. Davis, Chem. Rev., 118, 5265(2018).
21) S. A. Morris, G. P. M. Bignami, Y. Tian, M. Navarro, D. S. Firth, J. Čejka, P. S. Wheatley, D. M. Dawson, W. A. Slawinski, D. S. Wragg, R. E. Morris, S. E. Ashbrook, Nat. Chem., DOI: 10.1038/NCHEM.2761.
22) P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris, J. Čejka, Chem. Soc. Rev., 44, 7177(2015).
23) D. C. Calabro, J. C. Cheng, R. A. Crane, Jr., C. T. Kresge, S. S. Dhingra, M. A. Steckel, D. L. Stern, S. C. Weston, U. S. Patent 6049018(2000).
24) D. L. Dorset, S. C. Weston, S. S. Dhingra, J. Phys. Chem. B, 110, 2045(2006).
25) D. L. Dorset, G. J. Kennedy, K. G. Strohmaier, M. J. Diaz-Cabañas, F. Rey, A. Corma, J. Am. Chem. Soc., 128, 8862(2006).
26) S. Elomari, A. W. Burton, K. Ong, A. R. Pradhan, I. Y. Chan, Chem. Mater., 19, 5485(2007).
27) 窪田好浩,稲垣怜史,中澤直人,触媒,60, 247(2018).
28) S. I. Zones, S. -J. Hwang, M. M. Olmstead, S. J. Teat, A. Jackowski, A. W. Burton, C. Kim, J. Am. Chem. Soc., 122, 263(2000).
29) R. L. Lobo, M. E. Davis, Micropor. Mater., 3, 61(1994).
30) R. Bandyopadhyay, Y. Kubota, N. Sugimoto, Y. Fukushima, Y. Sugi, Micropor. Mesopor. Mater., 32, 81(1999); R. Bandyopadhyay, Y. Kubota, N. Sugimoto, Y. Fukushima, Y. Sugi, Micropor. Mesopor. Mater., 34, 347(2000).
31) Y. Kubota, Y. Koyama, T. Yamada, S. Inagaki, T. Tatsumi, Chem. Commun., 44, 6224(2008).
32) M. Sasaki, Y. Sato, Y. Tsuboi, S. Inagaki, Y. Kubota, ACS Catal., 4, 2653(2014).
33) S. Inagaki, K. Takechi, Y. Kubota, Chem.Commun., 46, 2662(2010).
34) S. Park, Y. Watanabe, Y. Nishita, T. Fukuoka, S. Inagaki, Y. Kubota, J. Catal., 319, 265(2014).
35) S. Inagaki, Y. Tsuboi, Y. Nishita, T. Syahylah, T. Wakihara, Y. Kubota, Chem. Eur. J., 19, 7780(2013).
36) N. Nakazawa, S. Inagaki, Y. Kubota, Adv. Porous Mater., 4, 219(2016).
37) J. G. Moscoso, D.-Y. Jan, U. S. Patent 7922997(2011).
38) M. Matsukata, M. Ogura, T. Osaki, P. R. H. P. Rao, M. Nomura, E. Kikuchi, Top. Catal., 9, 77(1999).
39) N. Nakazawa, T. Ikeda, N. Hiyoshi, Y. Yoshida, Q. Han, S. Inagaki, Y. Kubota, J. Am. Chem. Soc., 139, 7989(2017); Correction: N. Nakazawa, T. Ikeda, N. Hiyoshi, Y. Yoshida, Q. Han, S. Inagaki, Y. Kubota, J. Am. Chem. Soc., 139, 12102(2017).
40) N. Nakazawa, Y. Yoshida, S. Inagaki, Y. Kubota, Micropor. Mesopor. Mater., 280, 66(2019).
41) N. Nakazawa, S. Inagaki, Y. Kubota, Chem. Lett., 45, 919(2016).
42) T. Ikeda, S. Inagaki, T. Hanaoka, Y. Kubota, J. Phys. Chem. C, 114, 19641(2010).
43) Y. Kubota, K. Itabashi, S. Inagaki, Y. Nishita, R. Komatsu, Y. Tsuboi, S. Shinoda, T. Okubo, Chem. Mater., 26, 1250(2014).
44) G. Engelhardt, D. Michel, High-resolution solid-state NMR of silicates and zeolites, p. 149, John Wiley & Sons Ltd.(1987).
45) J. Klinowski, J. M. Thomas, C. A. Fyfe, G. C. Gobbi, Nature, 296, 533(1982).
46) C. W. Skeels, D. W. Breck, Proc. 6th International Zeolite Conference, Ed. By D. Olson, A. Bisio, p. 87, Butterworths, London(1984).
47) G. M. Johnson, P. J. Mead, S. E. Dann, M. T. Weller, J. Phys. Chem. B, 104, 1454(2000).