メタンからの合成ガス製造は,メタンの化学原料への転換法として今なお高い注目を集めている。メタンから合成ガスを得る反応として,CO2を原料としたドライリフォーミング反応(二酸化炭素改質,Dry reforming of methane; DRM),水蒸気を用いた水蒸気改質(Steam reforming of methane; SRM),酸素を用いた部分酸化改質(Partial oxidation of methane; POM)の3種類ならびにそれらを複合したデュアルリフォーミング(DRM+SRM),トリリフォーミング反応が積極的に研究されている1,2)。なかでも,DRMは,Table 1にまとめたように二酸化炭素を化学原料として用いることができる点,二酸化炭素を多く含む劣質な天然ガスならびにバイオガスを原料に対応できる点,得られる合成ガスの組成がH2/CO = 1に近く,ヒドロホルミル化,カルボニル化といった含酸素官能基を付与する反応,Fischer-Tropsch反応の原料として適しているといった利点を有する1–4)。他方で,商業化のためには,炭素析出ならびに反応温度での活性金属のシンタリングによる触媒劣化の解決が求められる。本稿では,特に活発に研究がされているNi触媒を中心に,触媒構造によるDRM反応の特性ならびに長期触媒活性の向上に向けた触媒構造開発の研究についてまとめたい。また,近年筆者らが取り組んできたシリケート種を経由した金属微粒子内包型ゼオライト触媒(バードケージ型触媒)合成についても報告する。
Table 1. メタンから合成ガスを製造する反応の比較1) |
先述のとおり,DRM反応とは,メタンとCO2から合成ガスを生成する反応である。
CH4+CO2⇄2CO+2H2 ΔH298K°=247 kJ/mol(1)
本反応は,著しい吸熱反応であることから650~1000°Cで反応が行われる。本反応はRh,Ru,Ptといった貴金属が高い活性を示す一方でNiやCoといった卑金属も活性を示すことが知られている。特にNiは比較的高い活性と安価であることから強い関心を集めている。一般に,貴金属触媒では炭素(コーク)析出が少ないのに対し,卑金属触媒上では多量の炭素析出が生じる。これは,貴金属触媒のほうが担体上に高分散で担持されるためであると考えられている2,3)。
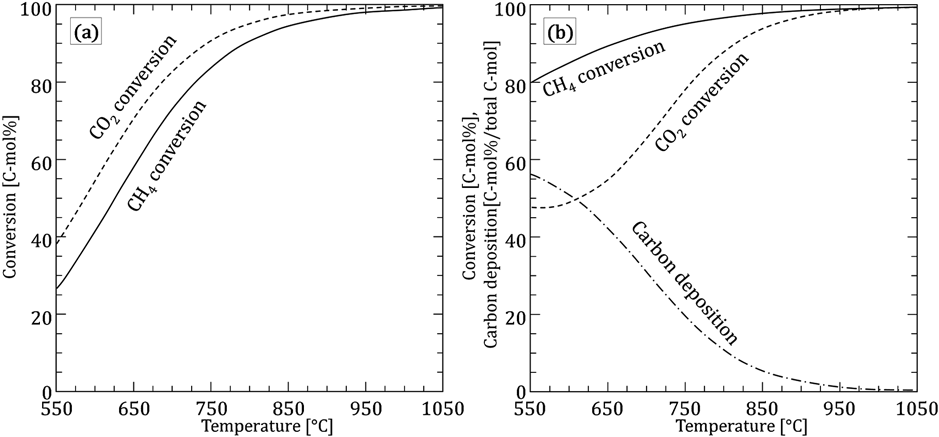
本反応は,非常に大きな吸熱を伴い,反応前後で分子数が増える反応であるため,高温・低圧条件が有利であることが知られている。実際にDRM反応の標準反応ギブズエネルギーは643°C以下では正の値をとるため,自発的にDRM反応を行うためにはそれ以上の高温が求められる。しかしながら,一般的なDRMの反応条件(600~850°C)においても熱力学平衡に基づく平衡転化率は最大でも95%にとどまり,熱力学平衡の制約を受ける。
DRM反応は,以下に示すメタン脱水素反応による水素生成と炭素のCO2ガス化反応(Reverse Boudouard reaction)の2つの反応に分けることができる。
CH4⇄C(s)+2H2 ΔH298K°=74.9 kJ/mol(2)
C(s)+CO2⇄2CO ΔH298K°=172 kJ/mol(3)
これらの反応では固体炭素が反応に関与するため,式(3)の炭素のCO2ガス化反応が式(2)のメタン脱水素反応に対して十分に遅い場合,固体炭素が蓄積する。Nikooら5)の熱力学計算によると,メタン脱水素,Boudouard反応に加えて,H2によるCOまたはCO2の還元反応による固体炭素の生成も起こり得る。また,反応系内にCO2とH2が共存するため,式(4)で表される逆水性ガスシフト反応(Reverse water gas shift reaction; RWGS)も副反応として進行する。
CO2+H2⇄CO+H2O ΔH298K°=41 kJ/mol(4)
とりわけ,RWGS反応はDRM反応に対して反応速度が非常に大きいことが知られており,DRM反応系内ではRWGS反応は速やかに平衡に達すると考えられている。DRM反応系では,H2Oの分圧が低く,式(4)は生成物側へと,すなわち,H2が減りCOが増える向きに反応が進む。これにより,炭素析出が無視できる反応条件では生成物のH2/CO比が1を下回る。
常圧,原料モル比CH4 : CO2=1 : 1でDRMの熱力学平衡計算を行った例をFig. 1に示す。炭素生成を考慮しない(式(1),(4)のみの熱力学平衡を考慮)場合,RWGSの影響によりCO2の平衡転化率がCH4の平衡転化率より大きくなり,H2/CO比が1より小さくなる。ただし,RWGSの影響は高温になるほど小さくなり,900°C以上ではH2/CO比はほぼ1に等しくなる。一方で,炭素生成を考慮した(式(2),(3),(4)の熱力学平衡を考慮)場合,低温ほど炭素生成の影響がより顕著にあらわれ,CH4の平衡転化率は炭素析出を考慮しない場合と比較して大きくなる。平衡計算では炭素析出量は650~850°Cで10% C-mol/total C-mol(原料中のCH4とCO2の総炭素数基準)以上を占めるため,特に低~中温域において炭素析出は無視できない問題となりやすい。また,平衡転化率および平衡炭素析出量は温度の他に,原料組成,反応圧力によって敏感に変化する5,6)。原料中のCO2/CH4比を大きくするほど,また反応圧力を小さくするほど,平衡炭素析出量は小さくなる傾向がある。
一方で,生成物と原料の分離は困難であるため,実用化を考えた場合,単通転化率をできるだけ高くすることが望まれる。したがって反応速度を上げるためにより高温での反応が必要となり,高い熱安定性を有する触媒が求められる。
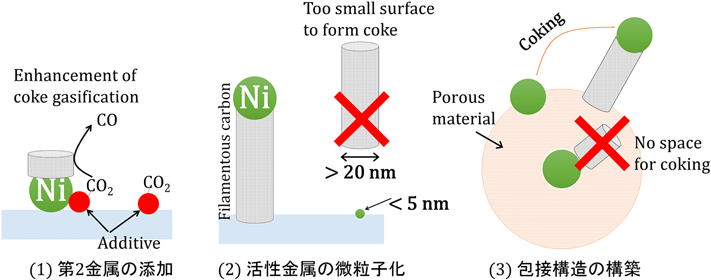
これまで述べてきたように,DRM反応においては低温ではNi触媒上への炭素析出,ならびに高温ではシンタリングによる触媒劣化が問題となる。そのため,炭素析出を抑制し,かつ,高い熱安定性を有する触媒が求められる。それに対し,様々な研究が行われてきた。炭素析出の抑制ならびに耐熱安定性の向上によるDRM活性劣化抑制の方針としては,①第2金属の添加,②活性金属の微粒子化,③金属包接構造の構築に大別することができる(Fig. 2)。本稿では,触媒構造に基づく炭素析出抑制および熱安定性向上に主眼をおくため,②活性金属の微粒子化,③金属包接構造の構築を詳しく採り上げ,①第2金属の添加については概説にとどめる。①第2金属の添加についての詳しい解説はAramouniのレビュー論文2)などを参照されたい。
3.1 第2金属添加
第2金属の添加は式(3)のCO2ガス化反応の促進による炭素析出の抑制を目的としている。第2金属添加の方針としては,(i)活性金属への第2金属添加による合金形成と(ii)担体への第2金属添加による担体表面の性状改質,金属担体間相互作用の強化がある。
活性金属への第2金属添加としては,Ni-貴金属系の二元金属触媒系が多く報告されている7,8)。貴金属担持による合金化では,貴金属上からNi上への水素のスピルオーバーによりNiを金属状態で安定化する,また,金属粒子が微粒子状態で安定化する効果が述べられている。また,Ni-Co二元金属系もDRM反応において炭素析出抑制能を持つと報告されている9)。
一方,Al2O3,SiO2などの金属酸化物担体への第2金属添加については,促進剤(プロモーター)としてアルカリ金属,アルカリ土類金属,Ce,Zr,Laなどの金属酸化物の使用が主に研究されている10–12)。アルカリ金属の担体表面に金属を添加することで,CO2をより多く吸着し固体炭素のガス化を促進する,アルカリ金属そのものが固体炭素のガス化反応の触媒として作用する10)といった効果が報告されている。しかし,過度な第2金属の添加はNi粒子表面上のメタン分解に寄与する活性点への吸着を引き起こし,DRM反応活性の低下を招く11)。
3.2 活性金属の微粒子化
活性金属の微粒子化は式(3)のCO2ガス化反応の促進に加えて,活性金属への炭素種の吸着を熱力学的に起こりにくくすることを狙いとしている。Ni粒子径が炭素析出量と相関があることは古くから報告されている。Bengaardら13)は,炭素析出はNi粒子のstep siteで前駆体が形成し,closed facet上で炭素種の重合が起こると報告している。さらに,DFT計算により炭素析出が起こるには80個以上,粒子径に換算して2.5 nm以上,のNiクラスターが必要との結果を得ており,実験からも粒子径が小さいほど炭素析出が起こりにくくなるとの知見を得ている。また,J. H. Kimら14)は,粒子径の異なるNiをAl2O3に担持した触媒を用い700°CでDRM反応を実施したところ,得られた繊維状の炭素に含まれるNi粒子径はいずれも7 nm程度あることから,Ni粒子径が7 nm以下のNi上には炭素析出が起こらないと結論付けている。この傾向はDuprezらの研究結果15)と合致している。このように,炭素析出を抑制するには金属粒子径を小さくすることが鍵となる。
Ni粒子を微粒子状態で担持する手法として,Z. Shangら16)はAtomic layer deposition(ALD)法を用い,多孔性のγ-Al2O3上に平均3.6 nmのNiを担持した例を報告している。ALD法とは,有機金属を加熱炉に供給し,熱分解反応により生成した金属元素を担持する手法である。担持したNiがγ-Al2O3細孔内でスピネル構造のNiAl2O4を形成することで高分散に担持されたために,還元時にNiが微粒子を形成したと考察している。700°Cでは炭素析出は観測されなかった一方で,850°CではNi粒子のシンタリングを伴う炭素析出が進行しており,炭素析出抑制効果は穏和な反応条件に制限される。また,Niを層状化合物の中に取り込むことで微粒子状態を実現した例も報告されている。例えば,Z. Bainら17)はナノチューブ状の層状化合物であるMg phyllosilicateのMg2+をNi2+に置き換えたNiMg phyllosilicateを触媒前駆体とし,650 °Cで還元することでナノチューブ内にNiを6~14 nm程度で担持することに成功している。しかし,750 °C,38 hにおけるDRM反応試験では,担体の構造変化を伴うNi粒子の凝集と炭素析出により,反応管閉塞を引き起こしたと報告している。
以上のように,微粒子状態で金属を担持した触媒の開発は進んでいるが,反応条件において微粒子状態を維持することが困難である。それに対し,次節に示す金属内包構造の構築による高温におけるシンタリングの物理的な抑制に関する研究が盛んに行われている。
3.3 金属内包構造の構築
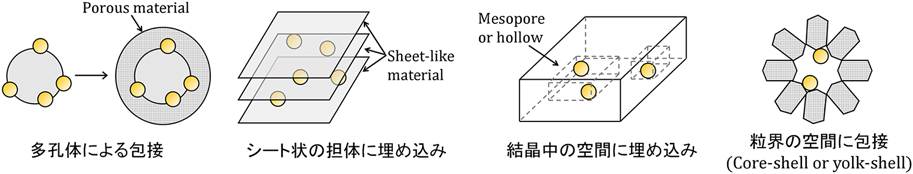
金属粒子を担体で内包した触媒構造では,担持金属の微粒子状態の維持に加えて,炭素析出が空間的に制限されるため,炭素析出の抑制につながると考えられている18,19)。アモルファスシリカ19–24),アルミナ20,25),ハイドロタルサイト26),ゼオライト8,27)といった担体が用いられており,種々の包接構造が提案されている(Fig. 3)。
J. W. Hanら20)は,粒子径を2.6~17.3 nmに制御したNiをSiO2上に含浸担持したNi/SiO2の周囲をSiO2,Al2O3,MgO,TiO2,ZrO2層でそれぞれ被覆することで,Ni粒子内包型触媒(Ni/SiO2@MOx)を調製し,DRM反応を実施した。SiO2包接Ni/SiO2触媒(Ni/SiO2@SiO2)では,Ni粒子径が小さいほどDRM活性は向上した一方,Ni粒子径に関係なく炭素析出が抑制されたことを見出している。また,Al2O3,MgO,TiO2による包接では粒子径を小さく維持できた一方,ZrO2ではNiを包接できず,Ni粒子のシンタリングが見られた。800°C反応後の炭素析出はNi/SiO2@ZrO2とNi/SiO2@Al2O3で確認され,前者はNi粒子径の増大に,後者は担体酸点上での副反応に起因したと結論付けている。同様の結果はBaktashら25)によっても報告されている。彼らはZ. Shangらとは逆にNi粒子上にAl2O3源をALDにより担持することで,Ni内包Al2O3触媒を調製した。DRM反応の結果,750°C,50 hの反応では優れた活性と炭素析出抑制能を示したが,850°C,50 hの反応後ではNi粒子径の増加は見られなかったものの炭素析出が確認され,Al2O3の酸点上での副反応の可能性を指摘している。さらに,Al2O3層が厚くなるにつれ,DRM反応速度が低下し,担体層での拡散抵抗が大きいと考察している。
X. Duら26)は,MgとAlを主とするハイドロタルサイトの調製時にNiを添加することで,Ni(OH)42−を取り込んだハイドロタルサイトを調製し,還元処理によりハイドロタルサイトのナノシート間に9~11 nmのNi粒子を高分散で埋め込んだ触媒を開発した。750°C,6 hのDRM活性試験の結果,わずかにナノチューブ状の炭素が析出したもののナノシート構造,Ni粒子径は大きく変わらず,高い活性,触媒安定性を示したと報告している。
J. Zhangら21)は市販のNiO粒子(10~20 nm)をアモルファスシリカで包接し,750°CでDRM反応を行い,炭素析出量が1.2 wt%に減少したと報告している。F. Wangら22,23)は5 nm程度のNi粒子を10 nm程度のアモルファスシリカで内包した触媒を油中水滴型エマルション場で調製した。750°CにおけるDRM反応に適用した結果,反応前後で包接構造,Ni粒子径に変化がなく,ほとんど炭素析出が起きなかったことを見出した。ただし,アモルファスシリカによる包接では,反応温度においてアモルファスシリカの細孔閉塞による緩やかな活性低下が報告されている。X. Zhaoら24)は,Ni内包アモルファスシリカ触媒に対し,Ceを加えることでNi分散性と耐熱安定性を維持できることを見出した。また,Z. Bainら17)は上述のNiMg-phyllosilicate触媒前駆体を10 nm程度のアモルファスシリカ層でコーティングすることで還元時ならびに凝集を抑制し,750°C,60 hのDRM反応で緩やかな活性低下があるものの炭素析出は全く確認されなかったことを報告している。
一方,酸点を持たないゼオライトでは,担体上での副反応がない点,耐熱性に優れておりより高温においても結晶構造を維持できる点から,より高温でのDRM反応に対しても適用できると期待される。C. Daiら8)は,Ni-Pt二元金属をSilicalite-1に含浸担持したのち,高濃度の構造規定剤溶液中で水熱合成処理を施すことで,金属粒子を内包した中空ゼオライトを調製した。調製した触媒は800°C,6 hのDRM反応後も微粒子状態を維持しており,また析出炭素もほとんど生成しないことを確認している。さらに,Niのみを用いて同様の手順で調製したNi内包中空ゼオライトにおいても,一部Ni粒子がゼオライト外に移動したことによる炭素析出があるものの,含浸法で調製したNi担持ゼオライトに比べて,安定した触媒活性と高い炭素析出抑制能を示した。
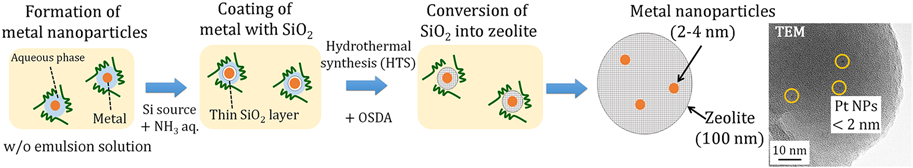
筆者らは,油中水滴型マイクロエマルション場で金属微粒子をアモルファスシリカで包接し,アモルファスシリカ層をゼオライトに転換することで,Pt微粒子内包ゼオライト(バードケージ型ゼオライト)を開発した(Fig. 4)27)。本調製法はゼオライトに後処理を施し,金属を包接する他の金属内包ゼオライト調製手法と順序が異なる点が特徴である。この触媒は2~4 nmの金属微粒子がゼオライト粒子内部にのみ存在する構造を有する。得られた触媒を用いてDRM反応を実施した結果,620°Cにおいて高いDRM活性を得られた。一方,670°C以上ではゼオライト結晶構造の崩壊に伴うPt粒子の凝集が進行した。ゼオライト結晶中に界面活性剤,有機溶媒等の不純物が取り込まれ,ゼオライト担体の熱安定性が低下したためであると結論付けている。
4. シリケートを経由したバードケージ型ゼオライトの調製
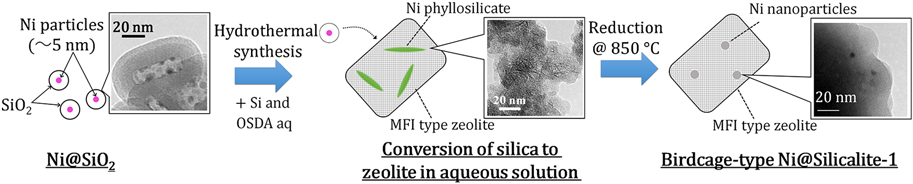
筆者らは近年,油中水滴型マイクロエマルション場を用いた金属微粒子内包バードケージ型ゼオライト触媒調製法に変わる調製法として,シリケート種を経由したバードケージゼオライト触媒調製法を開発した(Fig. 5)。本手法では,Ni微粒子内包アモルファスシリカ(Ni@SiO2)粉末28)をSi源と構造規定剤を含むゼオライト母液に添加し,Ni微粒子内包バードケージ型ゼオライト(Ni@Silicalite-1)に転換するものである。ゼオライト構造形成時にマイクロエマルション場を用いないため,有機溶媒や界面活性剤といった不純物を取り込まず,強固なネットワークを形成できるため,熱安定性の高いゼオライトが得られると期待される。
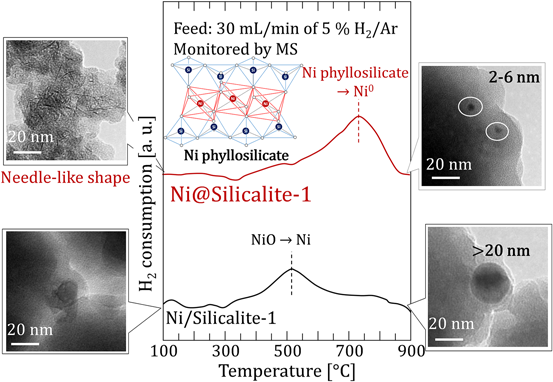
調製した触媒はXRD測定からMFI型結晶構造を有し,窒素吸着からミクロ孔の発達した細孔構造を有することが示された。一方で,原料となるNi種に対応する回折ピークは確認されなかった。また,TEM観察からは,針状の構造が確認された(Fig. 6)。本構造は,NiとSiからなる層状化合物であるNi phyllosilicate種29,30)に起因する。Ni phyllosilicateは水熱条件下,アルカリ溶液中で形成することから,Ni@Silicalite-1の水熱合成中において,NiはNi phyllosilicateを形成したと考えられる。実際に,H2-TPR(H2 temperature programmed reduction)測定からNi種の還元ピークは750°C付近に観察された(Fig. 6)。この温度はNiO粒子の還元温度である500°Cよりも200°C以上高く,文献で報告されているNi phyllosilicateの還元温度域29,30)と合致することから,Ni phyllosilicateが形成したと示唆される。本触媒を850°Cで還元すると針状の構造は消失し,2~4 nmのNi微粒子がゼオライト粒子内に確認された(Fig. 6)。一方,含浸法により調製したNi/Silicalite-1は同じ還元条件においてNi粒子が20 nm以上に凝集した。シンタリングが起こる苛酷な還元条件にもかかわらず微粒子状態が観察されたことから,Ni phyllosilicateはゼオライト一次粒子の粒界のようなゼオライト二次粒子内の制限された空間内に形成し,還元処理後もゼオライト二次粒子の内部に包接されたバードケージ構造が形成したと考えられる。以上から,Ni@SiO2をゼオライト母液に添加し,水熱処理を施すことで,Ni phyllosilicateを包接したゼオライトが形成し,水素還元処理によって,Ni微粒子内包バードケージ型ゼオライト触媒を得ることができた。
触媒構造の影響を確認するため,Ni微粒子をゼオライトに内包したNi@Silicalite-1,Ni微粒子をアモルファスシリカに内包したNi@SiO2, およびNi粒子が担体ゼオライトの内部だけでなく外表面にも存在するNi/Silicalite-1の3種類の触媒を用い,DRM反応活性を比較した。
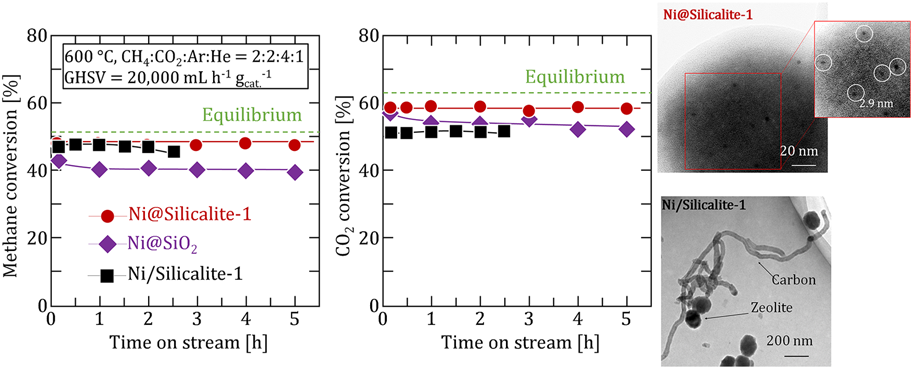
炭素析出が起こりやすい600°CにおけるDRM反応では,Fig. 7に示すように包接構造を有するNi@Silicalite-1とNi@SiO2は5 hにわたり安定した反応活性が得られた。特にH2/CO比はDRMとRWGSの平衡を考慮した平衡値よりもわずかに低い値を示しており,炭素析出が少ないことを示唆している。一方で,ゼオライト外表面にもNi粒子が存在するNi/Silicalite-1では転化率は2.5 hにわたり安定していたものの,H2/CO比が平衡値よりも大きくなり,炭素析出の可能性が示唆された。SEMおよびTEM観察から,Ni/Silicalite-1にのみ繊維状の炭素析出が見られた。一方,包接構造を有する触媒では,炭素析出ならびにNi粒子径の変化は見られなかった。加えて,反応後の触媒を用いたTG-DTAの結果,Ni/Silicalite-1のみ大きな重量減少が見られ,Ni@Silicalite-1およびNi@SiO2では重量変化がほとんど見られなかった。したがって,Niの包接構造により優れた炭素析出抑制能が発現したといえる。
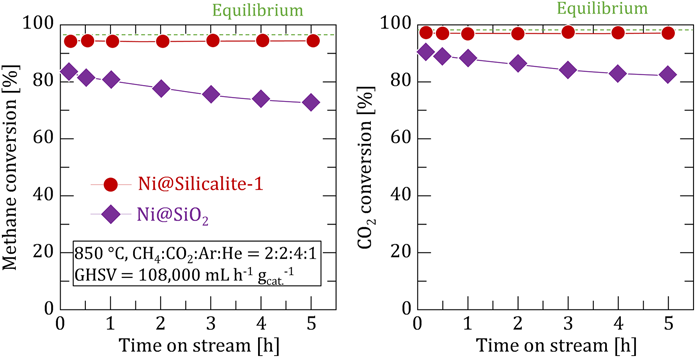
一方,熱安定性の評価のために850°CにおいてDRM反応を行った結果,Ni@Silicalite-1は5 hにわたり優れた活性と熱安定性を示したのに対し,Ni@SiO2では反応時間とともに活性低下が見られた(Fig. 8)。この触媒失活はアモルファスシリカ層の細孔構造の崩壊に起因したと考えられる。反応前後での窒素吸着によるBET表面積は,Ni@Silicalite-1ではほとんど変化が見られなかった(451 m2/g → 433 m2/g)のに対し,Ni@SiO2では172 m2/gから94 m2/gに著しく減少した。TEM観察から反応後のNi@SiO2は粒子径に変化が見られなかったことから,Ni@SiO2の活性低下はアモルファスシリカ層の熱的な細孔閉塞により,反応物がアクセスできなくなったためである。したがってNi@Silicalite-1はSilicalite-1の優れた熱安定性により,850°C反応においても活性低下を引き起こさなかったと結論した。
以上より,Ni@Silicalite-1はバードケージ構造に由来する活性金属の包接構造から炭素析出量を抑制することができ,かつ担体Silicalite-1の優れた熱安定性により高温での触媒劣化を抑制できるという優れた触媒性能を実現した。
Ni系触媒を中心に,DRM反応に対する触媒構造の影響をまとめ,バードケージ型触媒を用いたDRM反応の活性について述べてきた。炭素析出の抑制と高い熱安定性の実現にはNiの微粒子状態と包接構造を有するバードケージ構造が有効であると考えられ,DRM反応試験により実証した。この構造を活かした別の反応系への展開が期待される。
謝辞Acknowledgments
本研究の一部は,JSPS科研費JP17H03452および住友財団の助成を受けたものです。
引用文献References
1) B. Abdullah, N. A. A. Ghani, D.-V. N. Vo, J. Clean. Prod., 162, 170(2017).
2) N. A. K. Aramouni, J. G. Touma, B. A. Tarboush, J. Zeaiter, M. N. Ahmad, Renew. Sustain. Energy Rev., 82, 2570(2018).
3) S. Arora, R. Prasad, RSC Adv., 110, 108668(2016).
4) J.-M. Lavoie, Front. Chem, 11(2014).
5) M. K. Nikoo, N. A. S. Amin, Fuel Process. Technol., 92, 678(2011).
6) P. Cao, S. Adegbite, T. Wu, Energy Procedia, 105, 1864(2017).
7) M.García-Diéguez, I.S.Pieta, M.C.Herrera, M.A.Larrubia, L.J.Alemany, J. Catal., 270,136(2010).
8) C. Dai, S. Zhang, A. Zhang, C. Song, C. Shi, X. Guo, J. Mater. Chem. A, 3, 16461(2015).
9) D. San-José-Alonso, J. Juan-Juan, M. J. Illán-Gómez, M. C. Román-Martínez, Appl. Catal. A., 371, 54(2009).
10) J. Juan-Juan, M. C. Román-Martínez, M. J. Illán-Gómez, Appl. Catal. A: Gen., 264, 169(2004).
11) H. Liu, D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, P. D. Costa, M. E. Gálvez, Fuel, 182, 8(2016).
12) N. H. Elsayed, N. R.M. Roberts, B. Joseph, J. N. Kuhn, Appl. Catal. B: Environ., 179, 213(2015).
13) H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek, J. R. Rostrup-Nielsen, J. Catal., 209, 365(2002).
14) J.-H. Kim, D. J. Suh, T.-J. Park, K.-L. Kim, Appl. Catal. A: Gen., 197, 191(2000).
15) D. Duprez, M. C. Demicheli, P. Marecot, J. Barbier, O. A. Ferretti, E. N. Ponzi, J. Catal., 124, 324(1990).
16) Z. Shang, S. Li, L. Li, G. Liu, X. Liang, Appl. Catal. B: Environ., 201, 302(2017).
17) Z. Bian, I. Y. Suryawinata, S. Kawi, Appl. Catal. B: Environ., 195, 1(2016).
18) Z. Li, M. Li, Z. Bian, Y. Kathiraser, S. Kawi, Appl. Catal. B: Environ., 188, 324(2016).
19) A. J. Majewski, J. Wood, W. Bujalski, Int. J. Hydrog. Energy, 38, 14531(2013).
20) J. W. Han, J. S. Park, M. S. Choi, H. Li, Appl. Catal. B: Environ., 203, 625(2017).
21) J. Zhang, F. Li, Appl., Catal., B: Environ., 176, 513(2015).
22) F. Wang, B. Han, L. Zhang, L. Xu, H. Yu, W. Shi, Appl., Catal., B: Environ., 235, 26(2018).
23) F. Wang, L. Xu, W. Shi, J. CO2 Util., 16, 318(2016).
24) X. Zhao, H. Li, J. Zhang, L. Shi, D. Zhang, Int. J. Hydrog. Energy, 41, 2447(2016).
25) E. Baktash, P. Littlewood, R. Schomäcker, A. Thomas, P. C. Stair, Appl. Catal. B: Environ., 179, 122(2015).
26) X. Du, D. Zhang, L. Shi, R. Gao, J. Zhang, Nanoscale, 5, 2659(2013).
27) T. Kobayashi, F. Furuya, H. Fujitsuka, T. Tago, Chem. Eng. J., in press. https://doi.org/10.1016/j.cej.2018.10.140
28) S. Takenaka, H. Umebayashi, E. Tanabe, H. Matsune, M. Kishida, J. Catal., 245, 392(2007).
29) M. V. Sivaiah, S. Petit, M. F. Beaufort, D. Eyidi, J. Barrault, C. Batiot-Dupeyrat, S. Valange, Micropor. Mesopor. Mat., 140, 69(2011).
30) Z. Bian, S. Kawi, ChemCatChem, 10, 320(2018).