2. 非シリカ系のハイブリッド型メソポーラス材料の合成
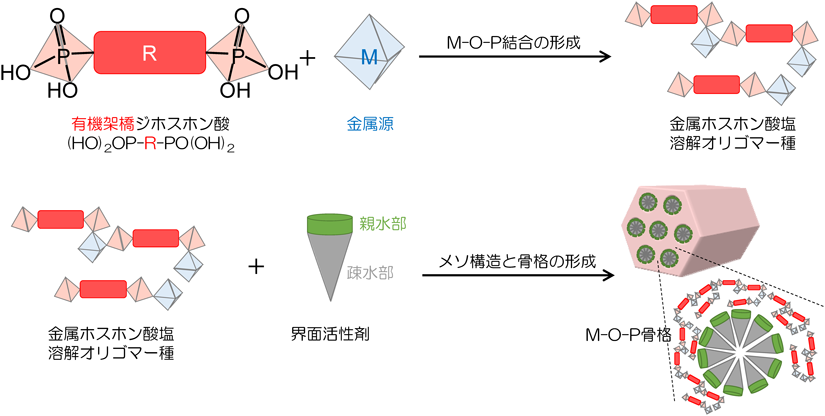
架橋ホスホン酸と金属源との反応を利用した非シリカ系メソポーラス材料の合成が2003年に初めて報告された21)。金属ホスホン酸塩からなる層状物質やゼオライト類縁化合物の報告例は多数あったが52–54),骨格内に有機基を導入した材料に関する報告例はほとんどなかった。M-O-P結合を含む溶解オリゴマー種と両親媒性有機分子(界面活性剤)との相互作用と協奏的組織化により生成するメソポーラスMOPは,従来の水熱合成法による層状構造やミクロポーラス構造を有する結晶性MOPとは全く異なる材料系であると理解できる。
2.1 メソスケールの構造設計
有機基の導入はメソポーラス材料の機能設計の最も有用な手法の一つであり,例えば,シランカップリング剤によるメソポーラスシリカの有機修飾と機能付与が有名である55–58)。2000年前後には,有機架橋部位を有するシラン系化合物(シルセスキオキサン)を利用したメソポーラス材料(PMO: Periodic Mesoporous Organosilica)の合成が報告されている59–64)。PMOの骨格内には有機基が組み込まれているが,骨格中の有機基がベンゼンなどの芳香族化合物になると規則的に配列することもある63,64)。しかしながら,有機架橋部位を有する化合物を出発原料としたアプローチが適用できたのはシリカのみである。機能設計の観点から,シリカ成分から化学的な機能は発現しないので,有機基の性質を利用した用途開発が進められている。
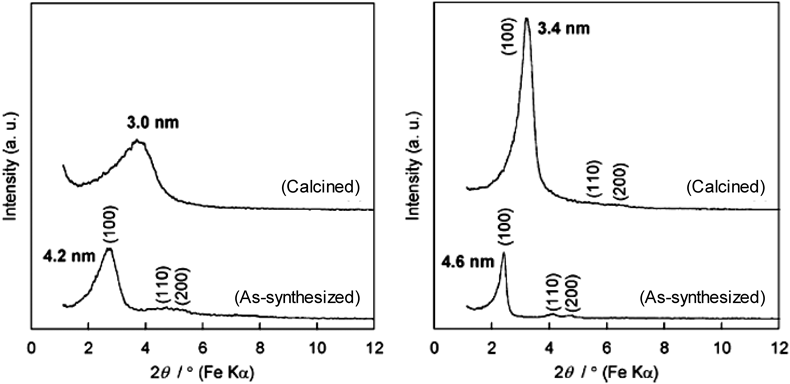
以上のような経緯から,我々は,世界で初めて,非シリカ系のハイブリッド型メソポーラス材料の合成法として,架橋ホスホン酸と金属源との反応による組成設計と両親媒性有機分子を利用したメソスケールでの構造設計の融合を提案した21)。ホスホン酸アルミニウム(AOP: Aluminum Organo-Phosphonate)のメソポーラス化に関する研究を通じて新たに提案した合成法の妥当性を実証した。図2に示したように,最初の論文ではメソポーラスAOPが塩基性条件で合成できることを示したが21,22),金属源を適切に選択すれば酸性条件でより構造規則性の高いメソポーラスAOPが合成できることを見出した22)。酸性条件下でアルキルトリメチルアンモニウム(CnTMA)系界面活性剤を用いて合成したメソポーラスAOPのX線回折(XRD)パターンからも明らかなように,界面活性剤除去後も二次元六方構造(空間群:p6 mm)に帰属可能な回折ピークが観測されている。
酸性条件での合成は,カチオン性のCnTMA系界面活性剤を利用した合成だけでなく,アルキルポリオキシエチレン(CnEOm)のような非イオン性界面活性剤やポリオキシエチレン–ポリオキシプロピレン–ポリオキシエチレン(EOnPOmEOn)系のトリブロック共重合体の利用も可能とした23)。各種メソポーラスAOPの透過型電子顕微鏡(TEM)観察の結果を図3にまとめたが,いずれの場合も二次元六方構造のメソポーラスAOPが得られていることを確認している。用いる界面活性剤のサイズに応じて,1.5 nm前後から10 nm程度の範囲で孔径制御できることも明らかにしている。
研究開始当初は,界面活性剤を抽出できず,低温焼成により除去していたため21–23),架橋有機基には耐熱性の高い単純なアルキル基(メチレン,エチレンなど)しか適用できていなかった。しかもP-C結合が一部切断されてしまうため,新たな界面活性剤の除去法の開発に迫られた。AOP骨格が水やエタノールの酸性溶液で簡単に溶解してしまうため,例えば,酸性溶液で処理して界面活性剤を抽出する方法は適用できなかった。別のグループからは,C16TMABrを用いてメチル基が結合したモノホスホン酸やエチレン基で架橋されたジホスホン酸からメソポーラス材料を合成したという報告もあったが,TEM観察からは虫食い状の低規則性メソ孔の存在しか示されていなかった24,25)。XRD測定の結果も低角度領域にブロードな回折ピークがひとつ観測されただけで,酢酸–エタノール溶液のような弱酸性の溶液で処理しただけで回折ピークはさらにブロード化していた。
種々の検討を重ねた結果,骨格内の有機基を完全に保持しつつ界面活性剤(Cn(EO)mとEOnPOmEOn)が除去できる方法として,脱水アセトン中で加熱処理して界面活性剤を分解する方法を見出すことができた26,27)。非常に湿度に敏感な方法であるが,テフロン内包型密閉容器にサンプルを入れて加熱処理するだけでCn(EO)mとEOnPOmEOnを分解,除去することができる。依然,メソポーラスAOP前駆物質からCnTMA系界面活性剤を除去する方法については開発の目途が立っていないが,アルミニウム以外の金属種,ホスホン酸チタン(TiOP),ホスホン酸バナジウム(VOP)およびホスホン酸鉄(FeOP)の合成で,塩酸–エタノール溶液を用いたC16TMABrの抽出が報告されているので28–30),CnTMA系界面活性剤の抽出に向けて引き続き検討していきたいと考えている。
2.2 マクロスケールの構造設計
階層的なメソポーラス/マクロポーラス構造を有するMOPが種々報告されている31–36)。AOPでは,比表面積は最大で154 m2 g−1とあまり大きくないが,有機架橋部が3級アミンであるホスホン酸からの合成で階層的な多孔体が得られている31)。界面活性剤にPluronic F127やPluronic P123を用い,水酸基を持つ有機架橋ホスホン酸(HEDP)から階層構造を有するTiOPが合成されており,比表面積は511 m2 g−1に達している32)。非イオン性ジブロック共重合体(EO30PO34)を用い,チタンテトラブトキシド由来のブタノールが生成するような環境でβ-シクロデキストリンやブチルアミンを添加して形成させたエマルジョンを利用することで,同様の階層構造を有するメソポーラスTiOPが得られている33,34)。エマルジョンはブチルアルコールのような疎水的な有機分子の存在によって形成されるので,界面活性剤を用いない単純な系でも階層的な多孔質構造を設計することもできる35,36)。
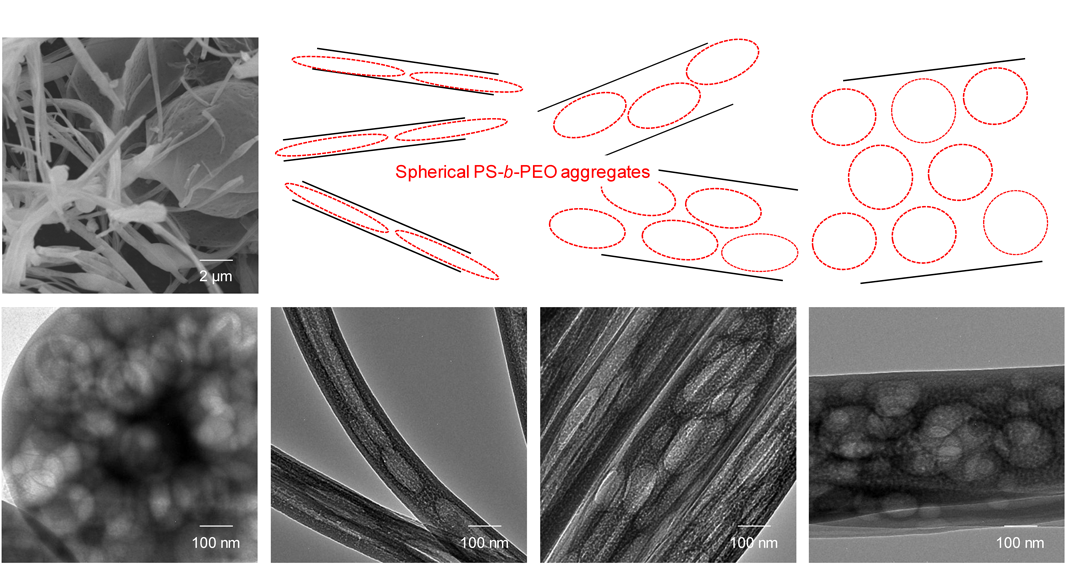
一般的に,規則的なマクロポーラス材料は粒径の揃ったポリマービーズやコロイダルシリカ粒子を鋳型として合成するが65–68),マクロポーラスMOPの合成にも同様の手法が利用されている37)。加えて,ポリスチレン−ポリエチレンオキシド(PS-b-PEO)ジブロック共重合体の球状コロイドを鋳型としたマクロ孔の導入も報告されている38,39)。PS-b-PEOの濃厚溶液に水を添加することで球状コロイドを形成させ,AOPの前駆溶液と混合してからスピンコート或いはスプレードライすることで,球状マクロ孔を内包する薄膜或いは粉体試料を得ることができる。PS-b-PEOは添加する水の量で溶解性が変化し,凝集体の大きさ(凝集体中のPS-b-PEOの数)を様々に変えられ,これによって導入される孔径が30 nmから200 nmの範囲で制御できることが確認されている。ただし,PS-b-PEOの量が多くなると前駆溶液の粘性が高くなるので,スプレードライによる粉体合成の場合には,図4の走査型電子顕微鏡(SEM)観察で見られたように,繊維状の試料が混在するようになる。また,PS-b-PEOの球状コロイドが柔軟性を有するため,TEM観察から,繊維状形態の内部ではその影響を受け,マクロ孔の形状が球状から楕円状へと大きく変化する様子が確認されている39)。
2.3 形態制御と溶媒揮発速度の重要性
界面活性剤を用いて合成されるメソポーラス材料は,粉体試料だけでなく,薄膜,ファイバー,ビーズ,モノリスのような多様な形態で得ることができる。例えば,薄膜化などの形態制御を行う合成法には,溶媒揮発(EISA: Evaporation-Induced Self-Assembly)プロセスが含まれている69–73)。均質な前駆溶液を調製した後,スピンコーティングなどの際に溶媒が揮発する過程で界面活性剤の濃度が上昇し自己集合が誘起されメソポーラス構造が形成されるというものである。先に述べたメソポーラスAOPの合成でも,プラスチックトレーに前駆溶液を流し入れ,室温で一晩,次いで50°Cで乾燥という穏やかな条件で溶媒を除去した結果として,構造規則性の高いサンプルを得ているため,溶媒揮発法を含む合成プロセスになっていると理解できる。
他方,トリブロック共重合体を用いたメソポーラスAOPの合成に関しては,球状粒子(粉体)及び薄膜としてサンプルを作成するため,透明な前駆溶液を調製し,それぞれスプレードライ及びスピンコートによる形態制御を試みた40–43)。スプレードライは噴霧温度によって溶媒の揮発速度を変えることができ,スピンコートは室温でしか操作できないが,スプレードライよりも溶媒の揮発速度は速いと考えて問題ない。噴霧乾燥する温度など,様々の合成条件を検討した結果,スプレードライでメソポーラスAOPを球状粒子として回収することが可能であった。この場合は低温(例えば110°C)でのスプレードライにより構造規則性のより高いメソポーラスAOP球状粒子を得ることができた43)。しかしながら,溶媒の揮発速度が速くなってしまうスピンコートでは,従来と同程度までメソスケールでの構造規則性を高めることは難しかった。以上の結果から,メソポーラスAOPの合成では,溶媒の揮発速度が生成する構造規則性に大きく影響しており,溶媒の揮発速度が遅い方が適していることが示唆されている。
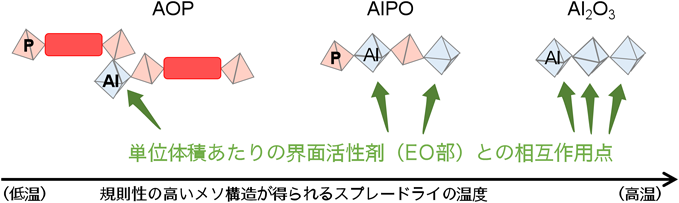
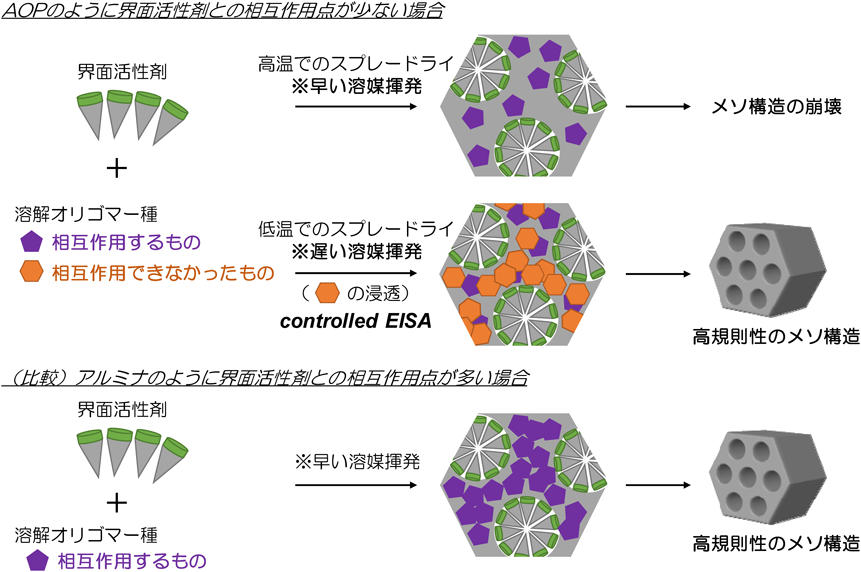
スプレードライを利用したメソポーラスアルミナ,メソポーラスシリカ,メソポーラスジルコニアといった無機酸化物の合成では,同じエタノール–水系の前駆溶液を使用していたが,高温(例えば400°C)のスプレードライで合成されていた。また,ホスホン酸アルミニウムと組成が近いメソポーラスリン酸アルミニウム(AlPO)でも,中低温(例えば170°C)のスプレードライでメソスケールの構造規則性が最も高く,それより高い温度でもメソ構造に由来するXRDのピークは明瞭に観測されていた。以上の結果をより正確に理解するため,溶媒の揮発速度に加えて,ハイブリッド骨格を形成する溶解オリゴマー種における界面活性剤との相互作用点の数が影響していることを考察した43)。図5に各種溶解オリゴマー種とEOnPOmEOnとの相互作用を模式的に示したが,溶解オリゴマー種はEOnPOmEOnのEO部と酸性の前駆溶液中に存在するH+を介して相互作用すると考えられる。AOPの溶解オリゴマー種は単位体積あたりの相互作用点となるアルミニウムのサイトがAlPOやアルミナよりも少ない。従って,図6にまとめたように,AOPの場合は,界面活性剤と相互作用できなかった溶解種も骨格内に取り込むためには,ゆっくりと溶媒を揮発させなければならない。これによりメソ構造体の骨格密度を高めることができ,構造安定性が高まると考えた。このような溶媒の揮発速度まで高度に制御したメソポーラス構造化技術のことを「controlled EISA」と命名した。
2.4 無機種と有機基の多様化
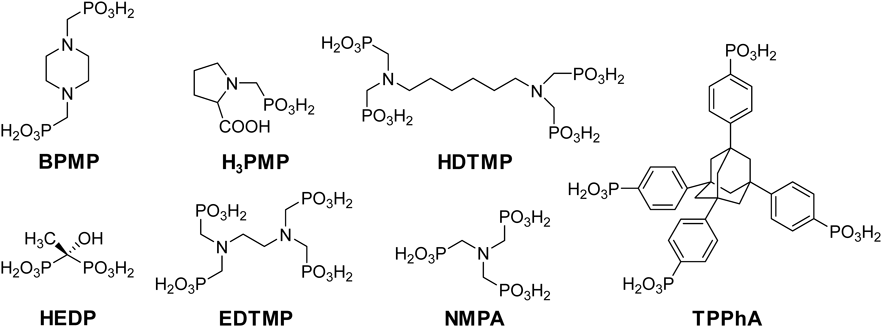
メソポーラスAOPの合成技術を基盤として,ジルコニウム(Zr),ニッケル(Ni),チタン(Ti),バナジウム(V)といった様々な金属種でも図7に挙げる有機架橋ホスホン酸を用いたメソポーラスMOPの合成が報告された。ドデシルスルホン酸ナトリウムを界面活性剤に用いて,p-ジメチルピペラジン構造を有する有機架橋ホスホン酸BPMPやL-プロリン構造を有するホスホン酸H3PMPからメソポーラスホスホン酸ジルコニウム(ZrOP)が合成された44,45)。ただし,これらの報告では,メソスケールの構造規則性の存在を裏付けるような分析結果は示されておらず,窒素吸脱着測定によるメソ孔の存在が示されているのみであった。ヘキサメチレンN,N,N’,N’-テトラキス(メチルホスホン酸)(HDTMP)を用いることで,ミクロ孔を有する結晶性の層状ホスホン酸ニッケルが合成された74)。この材料は結晶性の層状物質であり,ホスホン酸ニッケルシートが有機リンカーによって保持され,層間にミクロ孔が存在している。多様な層状ZrOPと同様に,骨格内に有機基が存在しているわけではない52,53)。
HEDPやEDTMPと金属源(四塩化チタン:TiCl4)をオートクレーブ中で加熱しながら生成物中に含まれる溶媒を除去することで,メソポーラスTiOPがモノリス状の試料として回収される46–49)。界面活性剤にはBrij56(C16EO10)とC16TMABrが用いられており,いずれの場合も二次元六方構造のメソポーラスTiOPが得られている。C16TMABrを用いた際には,立方構造(Ia-3d)のメソポーラスTiOPを得ることも可能であり,均一な球状粒子(400~500 nm)として回収されている49)。また,ミクロ孔を有する結晶性の細孔壁からなるメソポーラスMOP(M=Ti, Zr, V, Al)も合成できると主張されている50)。ただし,その根拠となっているTEM観察の結果は,結晶相を観察したとした格子縞が示されているが,金属の種類に関係なく似通ったXRDパターンしか示されていない。結晶性のラメラ構造のAOPとして報告したAOP-1に比べても51),固体31P MAS NMRの結果もブロードなピークしか検出できていない。
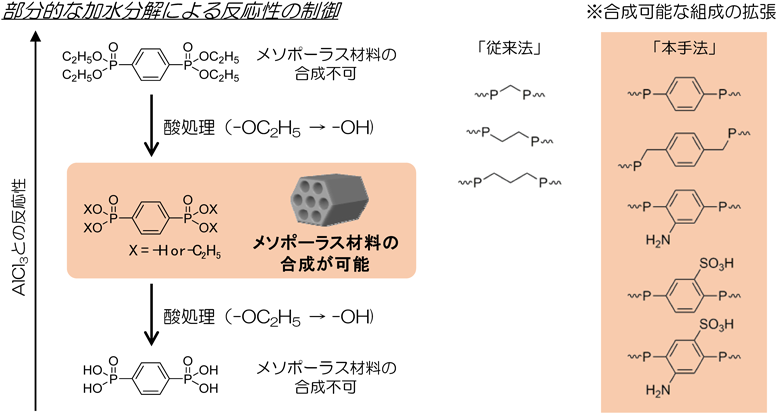
かさ高い有機基からなるTPPhAを用いても,界面活性剤なしで,メソポーラスVOPとTiOPが合成されている75,76)。その他にも,CnTMABr(n=14, 16, 18)を用い,有機架橋シラノレートホスホン酸とアルミニウム源を塩基性条件下で反応させることで二次元六方構造のホスホン酸アルミニウムシラノレートが合成されている77)。C16TMABrの存在下でNMPAと硝酸鉄(Ⅲ)および水酸化テトラメチルアンモニウム(TMAOH)を混合,水熱合成することで骨格が結晶化したメソポーラスFeOPが合成された29)。NMPAとメタバナジン酸アンモニウム(NH4VO3)およびTMAOHからもメソポーラスVOPが合成された30)。また,C16TMABrとアニオン性ポリマーであるポリアクリル酸(PAA)を併用することで,HEDPとの反応を経て,階層構造を有するTiOPも合成されている28)。
3. 非シリカ系のハイブリッド型メソポーラス材料の応用展開
メソポーラスAOPの水蒸気吸着特性は,界面活性剤を鋳型として合成されるメソポーラス材料の中でも特徴的である22)。リン酸アルミニウム(AOPの骨格に有機基を含まないもの)は吸着初期でのAlO4ユニットへの水の配位により高い親水性を示す78,79)。この性質はアルデヒドのような酸素原子を構造中に持つ分子の吸着にも効果的であった22)。メソポーラスAOPは有機架橋基の導入により若干疎水化するが,親水的な性質はある程度保持されており,リン酸アルミニウムの弱点であったメソポーラス構造の水に対する安定性を改善することができる。また,孔径が大きくなると多孔性AOPの水蒸気吸脱着挙動は,メソポーラスシリカよりもはるかに親水的な挙動を示し,珪藻のようなマクロ孔を有するシリカに似てくることもわかっている39)。
水酸基をもつHEDPを含むメソポーラスTiOPに塩化スルホン酸を反応させることで,スルホン酸基が導入でき,TiOP骨格自身に由来するイオン交換能(3.9 mmol g−1)を更に(+2.7 mmol g−1)増加させることに成功している46)。また,メソポーラスTiOPは,重金属(Cu2+)の吸着,ローダミンB(RhB)の光触媒による分解や液相中のCO2除去にも有用であることが示されている。エチレンジアミン基をもつEDTMPを含むメソポーラスTiOPはNa+やK+といったイオンの存在下でも金属錯体形成による重金属(Cu2+, Pb2+, Cd2+など)の捕捉に効果的であった51)。Cu2+の吸着後,焼成により酸化銅(Ⅱ)のナノ粒子をサンプル中に分散させることもでき,CO酸化活性を発現させることもできている48)。メソポーラスMOPは酸性,塩基性,中性の種々の化合物の高速液体クロマトグラフィー(HPLC)による分離にもその効果を発揮した50)。HDTMPを含むミクロ孔を有するNiOPは,金属カチオン(Cr3+, Pb2+, Hg2+, Cd2+)に対する高い吸着能と水素化ホウ素ナトリウム存在下でのニトロベンゼンの還元に高い触媒活性を示した74)。pH応答性のBPMPを含むメソポーラスZrOPは,腫瘍の光線療法でのアニオン光センサとして利用された44)。H3PMPで修飾したメソポーラスZrOPはリソザイムやパパインの吸着に利用された45)。NMPAを含むメソポーラスFeOPやメソポーラスVOPは,双方ともリチウムイオン電池(LIB)の活物質として良好なサイクル特性を示した29,30)。
引用文献References
1) S. Kitagawa, M. Kondo, Bull. Chem. Soc. Jpn., 71, 1739(1998).
2) H. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Nature, 402, 276(1999).
3) B. Chen, M. Eddaoudi, S. T. Hyde, M. O’Keeffe, O. M. Yaghi, Science, 291, 1021(2001).
4) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O. M. Yaghi, Science, 295, 469(2002).
5) O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi, J. Kim, Nature, 423, 705(2003).
6) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O’Keeffe, O. M. Yaghi, Science, 300, 1127(2003).
7) H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y.-B. Go, M. Eddaoudi, A. J. Matzger, M. O’Keeffe, O. M. Yaghi, Nature, 427, 523(2004).
8) J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard, O. M. Yaghi, Science, 309, 1350(2005).
9) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe, Y. Mita, Nature, 436, 238(2005).
10) S. T. Meek, J. A. Greathouse, M. D. Allendorf, Adv. Mater., 23, 249(2011).
11) N. Stock, S. Biswas, Chem. Rev., 112, 933(2012).
12) H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science, 341, 974(2013).
13) P. Silvia, S. M. F. Viela, J. P. C. Tome, F. A. Almedia Paz, Chem. Soc. Rev., 44, 6774(2015).
14) T.-Y. Ma, Z.-Y. Yuan, ChemSusChem, 4, 1407(2011).
15) T. Kimura, J. Nanosci. Nanotechnol., 13, 2461(2013).
16) Y.-P. Zhu, T.-Z. Ren, Z.-Y. Yuan, New. J. Chem., 38, 1905(2014).
17) Y.-P. Zhu, T.-Y. Ma, Y.-L. Liu, T.-Z. Ren, Z.-Y. Yuan, Inorg. Chem. Front., 1, 360(2014).
18) 木村辰雄,PHOSPHOROUS LETTER, 91, 20(2018).
19) T. Kimura, Angew. Chem. Int. Ed., 56, 13459(2017).
20) http://www.aist.go.jp/aist_j/press_release/pr2017/pr20171012/pr20171012.html
21) T. Kimura, Chem. Mater., 15, 3742(2003).
22) T. Kimura, Chem. Mater., 17, 337(2005).
23) T. Kimura, Chem. Mater., 17, 5521(2005).
24) J. E. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán, P. Amorós, Eur. J. Inorg. Chem., 1804(2004).
25) J. E. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán, P. Amorós, Chem. Mater., 16, 4359(2004).
26) T. Kimura, K. Kato, Microporous Mesoporous Mater., 101, 207(2007).
27) T. Kimura, K. Kato, J. Mater. Chem., 17, 559(2007).
28) H. Li, Y. Sun, Z.-Y. Yuan, Y.-P. Zhu, T.-Y. Ma, Angew. Chem. Int. Ed., 57, 3222(2018).
29) M. Pramanik, Y. Tsujimoto, V. Malgras, S. X. Dou, J. H. Kim, Y. Yamauchi, Chem. Mater., 27, 1082(2015).
30) P. Mei, M. Pramanik, J. Lee, Y. Ide, Z. A. Alothman, J. H. Kim, Y. Yamauchi, Chem. Eur. J., 23, 4344(2017).
31) T.-Y. Ma, X.-J. Zhang, Z.-Y. Yuan, J. Phys. Chem. C, 113, 12854(2009).
32) T.-Y. Ma, X.-Z. Lin, X.-J. Zhang, Z.-Y. Yuan, New J. Chem., 34, 1209(2010).
33) T.-Y. Ma, Z.-Y. Yuan, Eur. J. Inorg. Chem., 2941(2010).
34) T.-Y. Ma, L. Liu, Q.-F. Deng, X.-Z. Lin, and Z.-Y. Yuan, Chem. Commun., 47, 6015(2011).
35) X.-J. Zhang, T.-Y. Ma, Z.-Y. Yuan, Chem. Lett., 37, 746(2008).
36) T.-Y. Ma, X.-J. Zhang, Z.-Y. Yuan, Microporous Mesoporous Mater., 123, 234(2009).
37) T.-Y. Ma, X.-J. Zhang, G.-S. Shao, J.-L. Cao, Z.-Y. Yuan, J. Phys. Chem. C, 112, 3090(2008).
38) T. Kimura, Chem. Asian J., 6, 3236(2011).
39) T. Kimura, Y. Yamauchi, Langmuir, 28, 12901(2012).
40) T. Kimura, K. Kato, New J. Chem., 31, 1488(2007).
41) T. Kimura, K. Kato, Y. Yamauchi, Chem. Commun., 4938(2009).
42) T. Kimura, N. Suzuki, P. Gupta, Y. Yamauchi, Dalton Trans., 39, 5139(2010).
43) T. Kimura, Y. Yamauchi, Chem. Asian J., 8, 160(2013).
44) X. Shi, J. Li, Y. Tang, Q. Yang, J. Mater. Chem., 20, 6495(2010).
45) X. Shi, J. Liu, C. Li, Q. Yang, Inorg. Chem., 46, 7944(2007).
46) T.-Y. Ma, Z.-Y. Yuan, Chem. Commun., 46, 2325(2010).
47) T.-Y. Ma, X.-Z. Lin, Z.-Y. Yuan, J. Mater. Chem., 20, 7406(2010).
48) T.-Y. Ma, Z.-Y. Yuan, Dalton Trans., 39, 9570(2010).
49) T.-Y. Ma, X.-Z. Lin, Z.-Y. Yuan, Chem. Eur. J., 16, 8487(2010).
50) T-Y. Ma, H. Li, A.-N. Tang, Z.-Y. Yuan, Small, 7, 1827(2011).
51) T. Kimura, D. Nakashima, N. Miyamoto, Chem. Lett., 38, 916(2009).
52) A. Clearfield, Chem. Mater., 10, 2801(1998).
53) A. Clearfield, Metal Phosphonate Chemistry in Progress in Inorganic Chemistry, edited by K. D. Karlin, John Wiley, New York(1998), pp. 371–510.
54) K. Maeda, Microporous Mesoporous Mater., 73, 47(2004).
55) J.-W. Park, Y. J. Park, C.-H. Jun, Chem. Commun., 47, 4860(2011).
56) F. Hoffmann, M. Cornelius, J. Morell, M. Fröba, Angew. Chem. Int. Ed., 45, 3216(2006).
57) N. Linares, E. Serrano, M. Rico, A. M. Balu, E. Losada, R. Luque, J. García-Martínez, Chem. Commun., 47, 9024(2011).
58) J. G. Croissant, X. Cattoën, M. Wong Chi Man, J.-O. Durand, N. M. Khashab, Nanoscale, 7, 20318(2015).
59) S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna, O. Terasaki, J. Am. Chem. Soc., 121, 9611(1999).
60) T. Asefa, M. J. MacLachlan, N. Coombs, G. A. Ozin, Nature, 402, 867(1999).
61) J. Melde, B. T. Holland, C. F. Blanford, A. Stein, Chem. Mater., 11, 3302(1999).
62) M. P. Kapoor, S. Inagaki, Bull. Chem. Soc. Jpn., 79, 1463(2006).
63) S. Inagaki, S. Guan, T. Ohsuna, O. Terasaki, Nature, 416, 304(2002).
64) S. Fujita, S. Inagaki, Chem. Mater., 20, 891(2008).
65) B. T. Holland, C. F. Blanford, A. Stein, Science, 281, 538(1998).
66) J. E. G. J. Wijnhoven, W. L. Vos, Science, 281, 802(1998).
67) A. Imhof, D. J. Pine, Nature, 389, 948(1997).
68) A. Imhof, D. J. Pine, Adv. Mater., 10, 697(1998).
69) M. Ogawa, J. Am. Chem. Soc., 116, 7941(1994).
70) M. Ogawa, Chem. Commun., 1149(1996).
71) Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn, M. H. Huang, J. I. Zink, Nature, 389, 364(1997).
72) Y. Lu, H. Fan, A. Stump, T. L. Ward, T. Rieker, C. J. Brinker, Nature, 398, 223(1999).
73) H. Fan, Y. Lu, A. Stump, S. T. Reed, T. Baer, R. Schunk, V. Perez-Luna, G. P. López, C. J. Brinker, Nature, 405, 56(2000).
74) A. Dutta, A. K. Patra, A. Bhaumik, Microporous Mesoporous Mater., 155, 208(2012).
75) M. Vasylyev, R. Neumann, Chem. Mater., 18, 2781(2006).
76) M. V. Vasylyev, E. J. Wachtel, R. Popovitz-Biro, R. Neumann, Chem. Eur. J., 12, 3507(2006).
77) M. Otsu, T. Yamazaki, Y. Sasaki, K. Maeda, Chem. Lett., 39, 496(2010).
78) T. Kimura, Y. Sugahara, K. Kuroda, Microporous Mesoporous Mater., 22, 115(1998).
79) T. Kimura, Microporous Mesoporous Mater., 77, 97(2005).