2011年3月11日に発生した東日本大震災による福島第一原子力発電所事故では,津波により建屋内に大量の海水が流入し,また,事故直後に海水を炉心冷却水として原子炉内に注入,その後も,炉心冷却のために数百トン/日の注水が続けられ,放射性セシウム(Cs)等の放射性物質を高濃度(~106 Bq/cm3)で含む放射能高汚染水が短期間で大量に発生し,原発サイト内での汚染水保管場所の確保も困難な状態となっている。
問題となった放射能高汚染水は,主に放射性セシウム(137Cs, 134Cs)や放射性ストロンチウム(90Sr)を含む塩水系汚染水で,1979年3月に発生した米国スリーマイル島原発事故でのホウ酸系汚染水とは液性が大きく異なっている1,2)。
このため,ナトリウム(Na),カルシウム(Ca),マグネシウム(Mg)等の干渉物質の共存下で,CsやSr等の放射性核種を選択的に吸着除去できる高選択性吸着剤が求められ,また,処理しなければならない汚染水の量や核種の多さもあり,スリーマイル島原発事故に比べ,より困難な状況である。
スリーマイル島原発事故では,Cs吸着剤としてチャバサイト型ゼオライト(米国UOP社製IONSIV IE-96)が,Sr吸着剤としてA型ゼオライト(米国UOP社製IONSIV A-51)が使用された1,2)。福島第一原子力発電所事故発生後暫くは,Sr処理は行わず,Csのみを処理したが,Csの選択的分離システムには当初キュリオン社のゼオライト(ハーシェライト)やアレバ社のフェロシアン化物が使用された。その後,準国産の東芝製セシウム吸着装置(SARRYシステム)にはUOP社のチャバサイト型ゼオライト(IONSIV IE-96)と高性能のUOP社の結晶性シリコチタネート(IONSIV IE-911)が並行投入され,Cs処理に貢献した。
上述した,アレバ社システムで使用されたフェロシアン化物は,放射性Csを吸着した使用済みフェロシアン化物スラッジの長期安定保管が問題となっているが,このCsを吸着したフェロシアン化物に対し東北大学と共同で開発し,IRIDの委託研究としても実施したゼオライトを利用した安定固化法についても触れる。
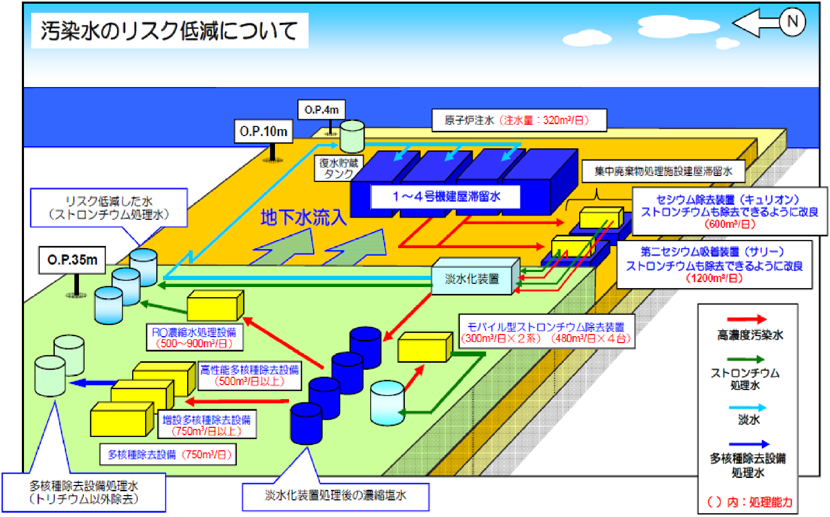
その後,汚染水処理のCs除去は一段落し,難易度の高い塩水中のSr除去に焦点が移った。Sr除去については,淡水系の米国スリーマイル島の汚染水では,UOP社のA型ゼオライト(IONSIV A-51)が効果を発揮したが,海水系の福島の汚染水には対応できず,効率的なSr除去剤が求められた。当時,セシウム吸着装置でCsを処理した汚染水は,福島第一原子力発電所内に設置されたタンクに貯蔵されていたが,タンクからの漏洩時の危険度低減のため,タンク内のCs以外のSr等の多核種を対象とした早期処理が求められ,多核種除去設備として多核種除去設備/ALPS,高性能多核種除去設備/HERO,増設多核種除去設備/増設ALPSの3システムが稼働した3,4)。多核種除去設備には,新たに開発されたSr吸着剤,ヨウ素(I)吸着剤が採用された。この汚染水処理のため開発された各種吸着剤は,その後設置されたSub-drainシステム等にも使用され,現在も継続している。
本稿では福島第一原子力発電所事故に対応すべく事故直後から開始したゼオライト系無機吸着剤をベースとした当社の各種吸着剤の開発について紹介する。
2. Sr吸着用のA-51JHPの開発と改良および量産
ゼオライトにイオン交換能力があることは広く知られており,洗剤にゼオライトを混ぜて水に溶けているCaやMgをイオン交換で捕集して水を軟化し,洗浄力を高める洗剤用ビルダーとして応用されている。また,ゼオライトには一般的なイオン交換樹脂の2~10倍以上のイオン交換容量があることも大きな魅力であり,ゼオライトの種類によりその金属毎のイオン交換の能力・選択性が異なっている。例えば,福島第一原子力発電所の汚染水対策で使用されているIE-96と呼ばれるチャバサイト型ゼオライトの選択性は,Cs>K>Na>Liの順序であり,Csに対して選択吸着性が高く,またA-51に代表されるA型ゼオライトは,Sr>Ca>Na>MgとSrの選択吸着性が高い5)。
UOP製チャバサイト型吸着剤には,Na形のIE-96の他に,K形のIE-95もあり,当初K形IE-95の利用も検討されたが,福島の汚染水がスリーマイル島原発事故に比べて難しい海水系であるから,より高性能のNa形IE-96を選択したと当時のUOPの担当者より連絡を受けている。
事故直後の3月下旬に電力中央研究所(CRIEPI)の方より,「今はとにかくCs値が高く,これをどうにかしないと現場に近づけない。まず,近づくためのCs処理を最優先で行った後に,必ずSrの処理の話が出てくるので,この時にはスリーマイル島原発事故で使用されたA-51を使う事になると考えている。」とのコメントを頂き,国難とも言える福島第一原子力発電所の除染を日本発の技術でとの方針から,国産Sr吸着用ゼオライトA-51J(Japan)の開発を開始した。
当社で開発したSr除染用ゼオライトA-51J(Japan),A-51JHP(Japan High Performance)は,スリーマイル島原発事故で実績のあるUOP社ゼオライトA-51をベース剤とし,その吸着性能を高め,海水系でも効果が発揮できるようにした改良品である6,7)。
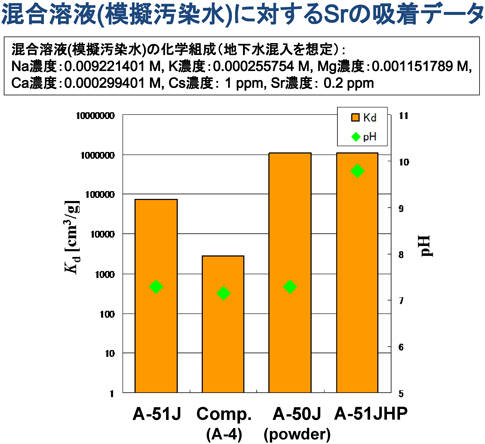
共同研究先である東北大学の三村教授にA-51Jの評価を依頼した処,図2に示すように,A-51Jの分配係数(Kd, cm3/g)は105と高く,UOP社や他社品のKd値が103程度であるのに対し,選択性が高く,差別化された製品であることが確認された。しかし,ゼオライト粉末と比較すると分配係数,吸着速度が共に低く,塩水系での使用には更なる改良が求められた。バインダーを含有する成形体はゼオライト含有量100%の粉末に対し,分配係数,吸着速度で劣るのは自明だが,東北大学と共同でその差をつめる方法を模索した結果,成形体を高ゼオライト化すれば汚染水とゼオライトの接触効率が向上するのではないかと考え,高ゼオライト化品を試作した。その結果,Kd値は105から106へ更に向上し,粉末に匹敵する分配係数,吸着速度が得られた。この評価結果をもとに,日本製A-51Jを高ゼオライト化し,ゼオライト成形体でありながら,塩水中でも有効な,粉末と同等のSr分配係数,吸着速度をもつA-51JHPを開発した。
本改良品はA-51JHPとして,三重県からのオンリーワン企業育成技術開発支援補助金の交付を受けてユニオン昭和(株)四日市工場の中に設置されている高ゼオライト化設備を利用して生産され,福島第一原子力発電所の汚染水処理設備に供給されている。
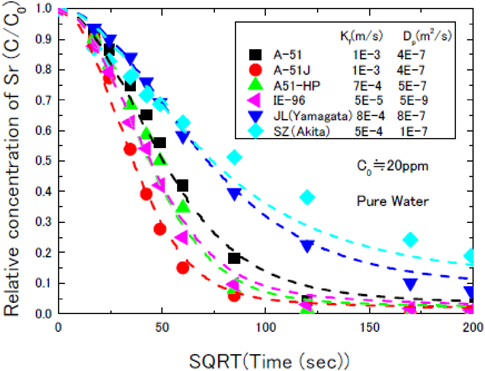
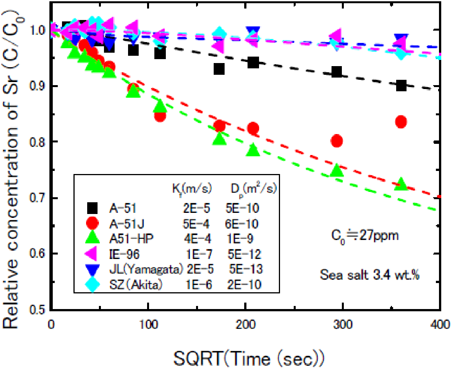
電力中央研究所により行われた純水系と海水系における,天然ゼオライト2種(JL:山形産,SZ:秋田産)とチャバサイト系ゼオライト(IONSIV IE-96)とA型ゼオライト3種(A-51, A-51J, A-51JHP)の吸着率の比較結果を図3および図4に示す。これらの図の比較から明らかな様に,いずれのゼオライトにおいても海水中で吸着速度が低下する傾向が認められるが,国産化A-51J, A-51JHPはその吸着速度の低下率が小さく,特に,海水系での使用を目的に開発されたA-51JHPは他ゼオライトに比し吸着速度が大きいことが確認された。本評価結果は,電力中央研究所からA-51J,A-51JHPは海水中でもSr吸着性能が高いと日本原子力学会で報告されている8)。
更に,実際に汚染水処理に使用される吸着剤には耐放射線性も要求される。
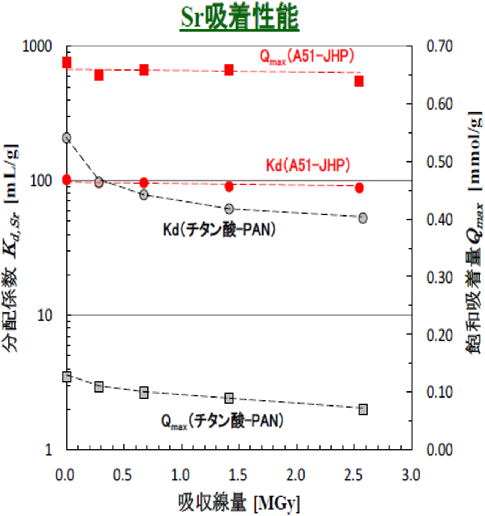
図5に,東北大学/日本原子力研究開発機構(JAEA)/当社の共同研究で実施されたγ線照射試験による耐放射線性の評価結果を示す。図から明らかな様に,チタン酸-PANの様に,バインダー成分に高分子を含む吸着剤は,吸収線量の増加によりKd(分配係数),Qmax (吸着容量)ともに低下する傾向が認められるが,無機系のゼオライト吸着剤は吸収線量の増加によるKdおよびQmaxの低下がほとんど認められず高い耐放射線性を有することが確認されている。このγ線照射試験については,使用時の劣化の他に,廃吸着剤貯蔵時の水素生成についても日本原子力研究開発機構等で各種吸着剤について比較検討がなされており,使用済み吸着剤の長期にわたる安定保管についても検討が続けられている。
福島第一発電所の港湾内の除染についても2013年に電力中央研究所,昭和環境システム社(現,ヴェオリア・ジェネッツ社)と共同で「浮遊式ゼオライトカラムとカラム再生プラントによる海水浄化技術」を検討・提案した。港湾開渠内は閉鎖されておらず,約16万立方メートルの容量があり,シルトフェンスで囲まれた開渠の部分の放射能とシルトフェンスの外の放射能を比較すると,開渠内のCsに関しては,2013年当時,ほぼ告示濃度以下になっていたが,Srに関しては,告示濃度より高く推移していることが確認されていた。
まず,スリーマイル事故や東芝製セシウム吸着装置(SARRYシステム)で使用されたUOP製IE-96や日本産天然ゼオライトについて海水中での吸着試験やカラム試験による評価を行いSrの吸着性能については,A-51系ゼオライトが最も高いことを確認した。
海水中には元々5 ppm程度のSrが存在するので,Srが計8 ppm存在する場合,A-51Jの吸着容量から1 gのゼオライトに対して,1.1×10−2 mmolのSrが吸着できることが分かり,90Sr濃度が告示濃度の30 Bq/Lに低下するまでには,A-51Jゼオライトで約3,000 ton,A-51JHPゼオライトで約1,800 tonもの大量のゼオライトが必要となることが判明した。
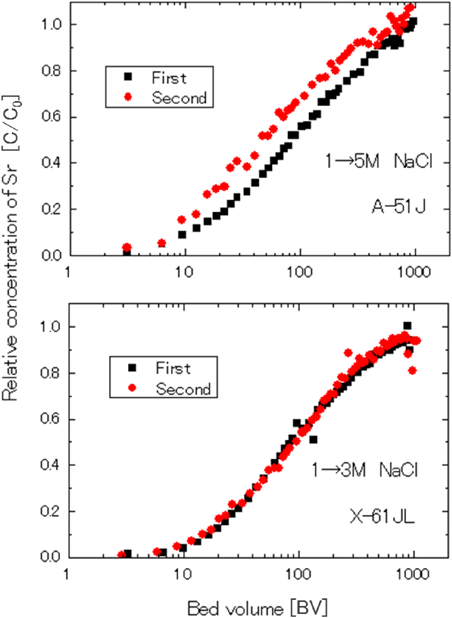
そこで,ゼオライトの使用量を減らすためゼオライトの再生使用の検討を行った。まず,ゼオライトを充填したカラムの上方から,Srを添加した人工海水を通液して,飽和吸着までのカラムの破過曲線を確認した後,1~5 mol/LのNaCl水溶液をカラムの下方から逆洗することでSrを脱離させて吸着剤を再生した。再生後に,再度,Srを添加した人工海水を通液して,カラムの破過曲線を確認する試験を繰り返した。A-51JのNaClによる再生試験では,吸着にくらべ,脱離しにくく,Na濃度を上げると高い濃度のSr溶液として回収はできたが,Srの脱離の総量はほとんど変わらなかった。更に吸着試験を継続すると,図6に示す様に,2回再生の破過曲線は1回再生の破過曲線より破過が早くなり,この傾向は再生液のNa濃度を高くしたり,Na濃度を変えても改善することはなかった。
次に,A-51系では期待した結果が得られなかったので,X型ゼオライトであるX-61JLの検証を行った。X-61JLを同様に再生した場合は,2回再生の破過曲線は1回再生の破過曲線と同等になり,再生を繰り返しても,吸着性能は低下せず,ほぼ100%再生できることが確認された。港湾除染での再生吸着剤としては,X-61JLが適しているとして,X-61JLによる再生吸着システムを提案した。
汚染水のCs除去が一段落した後,難易度の高い塩水中のSr除去に焦点が移った。2015年初めにはCs吸着装置によりCs除去処理を行った50万トン程度のCs処理水がタンクに貯蔵されていた。本Cs処理水はCs以外の放射性核種が未処理であったが,貯蔵汚染水漏洩時の危険度の低減目的で,図1に示す様に,多核種除去設備として3システム(多核種除去設備/ALPS,高性能多核種除去設備/HERO,増設多核種除去設備/増設ALPS)が当時稼働した。稼働後2015年5月末に,Sr濃度は1/1000程度に低減されており,東京電力はタンク内のCs以外の多核種除染処理がほぼ完了したと発表している。
本稿でCs用にはチャバサイトが,Sr用にはA型ゼオライトが有用と述べてきた様に,放射性核種除去には各核種に適した吸着剤があり,多核種の処理の際には,ガス分離の世界でよく使われる複数の吸着剤を吸着塔に充填して使用する,「パーシャルローディング」の手法が一般的であった。
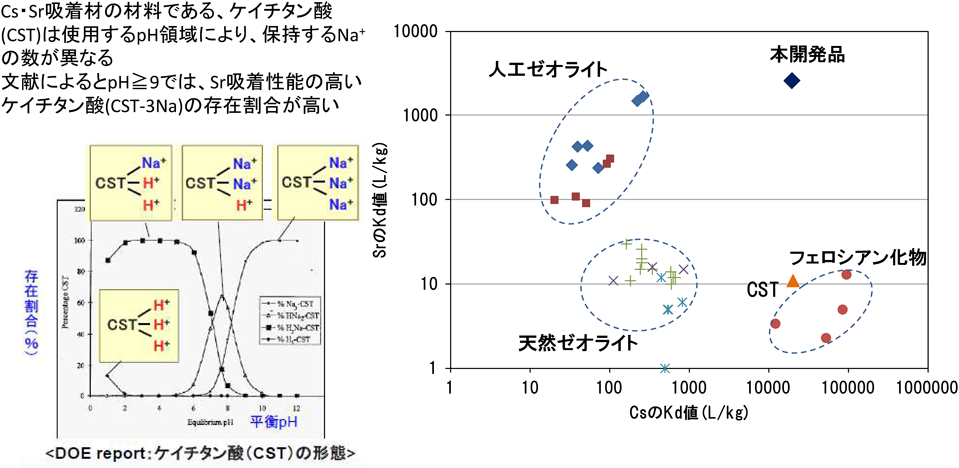
これに対し,高性能多核種処理設備には単一の吸着剤でCsとSrを同時に吸着剤するCST型IE-911が使用されていると東京電力のHPにも掲載されおり,本同時吸着剤は2013年に開発された吸着剤として報告されている10)。従来より高性能Cs吸着剤として準国産の東芝製セシウム吸着装置(SARRYシステム)にも使用されてきたUOP製結晶性シリコチタネート(CST:UOP製IE-911)は,使用するpH領域により保持するNaの数が異なり,pHが9以上の領域ではSr吸着能の高い3Na形ケイチタン酸(CST-3Na)の割合が高くなる(図7)。
本同時吸着剤は本性質を応用したもので,CSTを苛性ソーダ等で表面処理することで,CSTのCs吸着性能を損なうことなくSr吸着性能が付与されていると報告されている。
5. ヨウ素吸着用のイオン交換型銀ゼオライトの開発
従来ヨウ素吸着剤としては,活性炭に銀(Ag)を添着した吸着剤が提案されていた。これは,活性炭上に添着したAgと汚染水中に存在するヨウ素(I)の反応により生成するAgIの溶解度積が小さいことを利用したヨウ素吸着剤である。しかし,ベース剤の活性炭は耐放射線性に問題があり,電力中央研究所の要請により新たなゼオライト吸着剤の開発を開始した。
ヨウ素選択吸着性の高い金属イオンを選定するため,ゼオライトにIと難溶性沈殿を生成する各種金属イオンを交換吸着させIの吸着性を評価し,Iを高効率で吸着する金属イオンとしてAgを選択した。前述の活性炭に付着しているAgはイオン交換ではなく,表面に添着された金属銀の形態であり,これと同じくゼオライトの表面にAgを添着したゼオライトがHgSIVTMとしてUOP社から製造販売されている。これは,天然ガス中に含まれる水銀をゼオライト上に添着されたAgとアマルガムを生成させる事によりガスから除去するもので,当初このHgSIVTMの利用も検討した。しかし,イオン交換で添加している銀以外の,ゼオライト表面に添着された金属銀は,保管状況によって色調が変化し,色調が変化するとIとの反応性が低下し,ヨウ素吸着性能に影響し,安定した性能の吸着剤にならないことが判明した。そこで,添着ではなくイオン交換でAgをゼオライトに添加し,銀添加量を高めた銀ゼオライトを開発した。
図8は開発されたイオン交換法による銀ゼオライトの,ゼオライト上のAgとIを吸着させた後のAgIのEDX分析結果を示す。
イオン交換で導入したAgがIと反応して,Iをゼオライト内に固定化していることが確認できる。
6. 多核種を同時に吸着するハイブリッド化ゼオライト吸着剤の開発
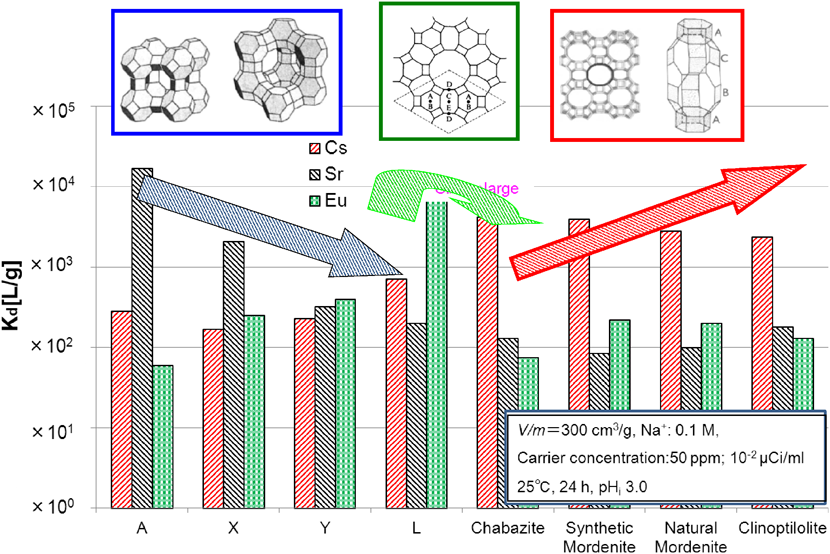
ゼオライトは,その構造とSi/Al比により核種選択性が大きく異なる。図9は各種ゼオライトのCs,SrおよびEuの分配係数値の比較を示す。
図9から明らかな様に,Csに対してはスリーマイル島原発事故でも使用され,SARRYシステムでも使用されている提灯型のチャバサイト及びトンネル状孔路を有するモルデナイト,クリノプチロライトが高い選択性を有する。Srに対しては,スリーマイル島原発事故でも使用され,Si/Al比が小さく,かつ三次元かご型構造を有するA型ゼオライトが高い選択性を有し,A型ゼオライト>X型ゼオライト>チャバサイト>モルデナイトとなる。また,L型ゼオライトは孔路径が大きいトンネルとケージの両方を有する構造であり,3価カチオンのEuやAmに高い吸着性を有している。
核種選択性が異なるゼオライトを混合造粒してハイブリッド化することにより,多核種に選択性を有する吸着剤の開発が可能である。一例として,SrおよびCoに選択性を有するA型およびX型ゼオライトと,Csに選択性を有するモルデナイトに,バインダー(造粒剤)を加えて混合焼成し,ハイブリッド剤として,AMXα(A型ゼオライト : モルデナイト : X型ゼオライト比,1 : 1 : 1)およびAMXβ(同,0.5 : 1 : 0.5)が開発されている(表1)。
表1. 使用したゼオライトと混合造粒試料の仕様| ベース剤 | ゼオライト種 | 粒径(mm) | 製造元 |
|---|
| A-51JHP | A Zeolite | 0.5~1.0 | ユニオン昭和 |
| 2460# | Mordenite | 0.5~1.0 | 新東北化学 |
| X-61JL | X Zeolite | 0.5~1.0 | ユニオン昭和 |
| AMXα | 三種混合 | 0.5~1.0 | ユニオン昭和 |
| AMXβ | 三種混合 | 0.5~1.0 | ユニオン昭和 |
AMX造粒試料の,純水系および海水系でのSr吸着率および平衡pHを比較した結果を,表2に示す。1/10海水では純水とほとんど変わらないSr吸着性能である。いずれの試料も,1/2海水では80%程度,海水系で60%程度に低下した。AおよびX型ゼオライト含有率が高いAMXαは,AMXβより高いSr吸着率を示した。試験後の平衡pHはAMXβが最も高いが,Srの加水分解が顕著となるpH 12よりも低い。
表2. 混合造粒試料のSr吸着率とpH( )内pH| Sr吸着剤 | 純水 | 海水1/10 | 海水1/2 | 海水 |
|---|
| AMXα | 98.7 (9.17) | 97.2 (7.89) | 84.9 (8.27) | 65.2 (8.35) |
| AMXβ | 99.6 (10.28) | 95.8 (8.83) | 75.8 (8.57) | 57.5 (8.68) |
AMX造粒試料の,純水系および海水系でのCo吸着率および平衡pHを比較した結果を,表3に示す。1/10海水では純水とほとんど変わらないCo吸着率を示した。単純配合のAMXαのCo吸着率は,1/2海水で70%,海水で47%にまで低下した。一方,いずれの条件下でも,AMXβは極めて高いCo吸着率を示した。平衡pHの上昇が大きいことから,Coの加水分解の影響が大きいと考えられる。
表3. 混合造粒試料のCo吸着率とpH( )内pH| Co吸着剤 | 純水 | 海水1/10 | 海水1/2 | 海水 |
|---|
| AMXα | 98.2 (9.39) | 96.2 (8.05) | 71.6 (6.31) | 46.6 (8.47) |
| AMXβ | 94.6 (10.03) | 99.2 (8.87) | 95.8 (7.58) | 89.6 (8.84) |
以上の結果から,海水濃度が1/10程度まで薄まれば,混合造粒吸着剤をセシウム吸着装置処理水の後段に設置し,Cs, Sr, Coの一括除染に適用できる可能性がある。循環注水冷却により汚染水の塩分除去が進行しており,ゼオライト系吸着剤の適用可能性は広がっている。各種ゼオライトの多様な核種選択性を利用して,混合造粒吸着剤の多様化及び最適化を図るとともに,多核種の吸着特性をより詳細に明らかにすることが望まれる。
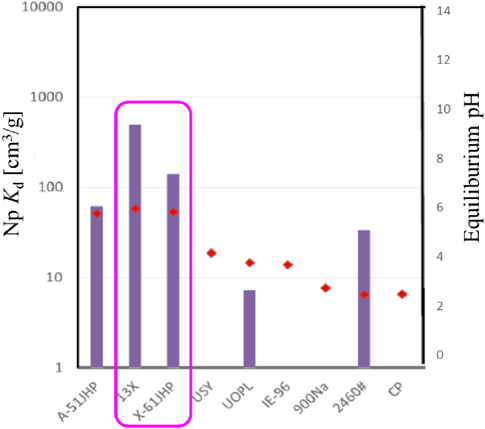
福島第一原子力発電所事故での高汚染水中に存在するアクチノイドの吸着剤への吸着挙動については明確な解析がなされていない。将来的には,燃料デブリ取り出し時に,アクチノイドの溶出および除染が重要な課題と考え,東北大学多元研と協力して,各種ゼオライトに対するU(VI),Am(III),Np(V)の分配特性に関して,Kdと平衡pHとの関係を調べ,ゼオライト構造,加水分解pH,化学形の観点からその核種選択性を評価した11,12)。
図10にNa溶液中での各種ゼオライトへのNp吸着評価を,図11に30%海水中での各種ゼオライトへのNp吸着評価を示す。Npの吸着に対しては,0.1 M NaCl-10−3M HCl共存下でのKd値はX型ゼオライトで最も高く,102 cm3/g以上の高い値が得られた。
U(VI)およびAm(III)は,酸性側でイオン交換吸着,中性で加水分解種の器壁吸着・沈降,アルカリ側では陰イオン形となりコロイド沈降することが確認された。Uの吸着については,中性では粒子表面に加水分解種の吸着による濃縮層が生成し,UのKdとAl含有率には相関があり,A型ゼオライトへのKdが高い。Amの吸着性能は,pHに大きく依存し,pH上昇に伴いKd値が増大し,構造(孔路径)による影響が大きく,孔路径が大きいL型ゼオライトが高い選択性を示すことが判明した。
前述の様に,ゼオライトの種類により,アクチノイドの吸着性に差が見られ,今後更に研究を続け,吸着速度,共存イオン濃度の影響,吸着機構を明らかにする予定である。
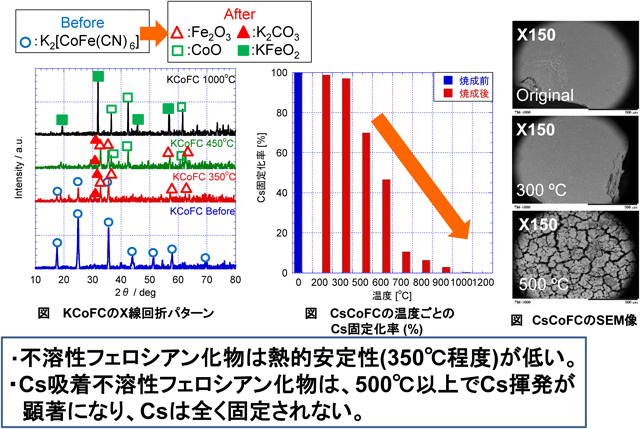
前述のアレバ社のシステムにも利用された不溶性フェロシアン化物は,Cs吸着後,放射性Cs吸着フェロシアン化物廃スラッジとして一時的に保管されている。しかし137Csの放射能は半減するのに30年かかり,廃スラッジのまま保管しておくと,崩壊熱でフェロシアン化物が分解し,捕捉されたCsが再度,外部飛散したり,シアンガスの発生が懸念されるため,Csを安定的に長期保管する方策が求められている。図12にCsを飽和吸着した不溶性フェロシアン化物の加熱時のCs固定化率(%)の変化を示す。Csを吸着した不溶性フェロシアン化物を加熱すると,300°C付近からフェロシアン化物の分解が始まりCsの保持力が低下し,500°C以上でCsの揮発が顕著になり,1,000°C以上ではCsは全て揮発してしまう。
当社は東北大学との共同研究で,Csを飽和吸着した不溶性フェロシアン化物をゼオライトと一緒に混合・加熱処理することにより,不溶性フェロシアン化物の熱分解により揮発したCsが高比表面積のゼオライトにトラップされ,Csが揮発することなく,安定固化出来ることを見出した13–16)。図13は,Csを吸着した不溶性フェロシアン化物を各種ゼオライトと混合・加熱処理した際のCs固定化率を示す。不溶性フェロシアン化物単独では図12に示した様に,Csは全く固定化されないが,ゼオライトと混合・加熱すればCsが安定固化されることがわかる。
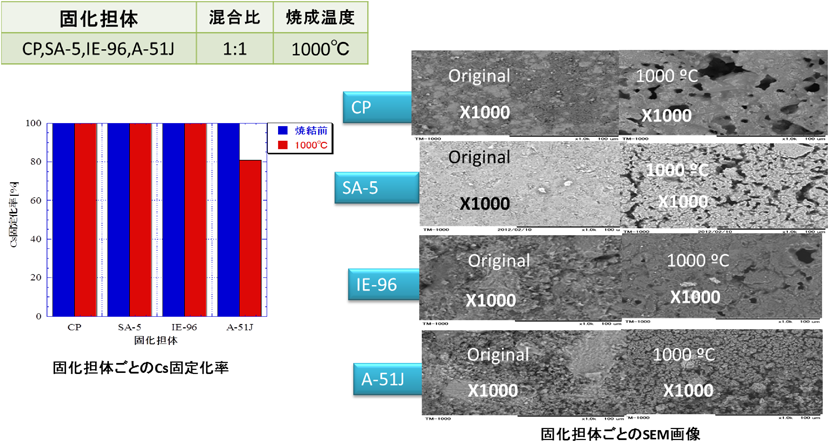
このように,ゼオライトはCs, Sr等に対して高いイオン交換能力を持つばかりでなく,高温での優れたCsガスのトラップ機能,安定固化を可能とする自己焼結機能を有していることが確認され,本技術を応用すれば,アレバ社システムによるフェロシアン化物スラッジを東芝製セシウム吸着装置(SARRYシステム)で使用したゼオライトで安定固化することも可能である。
本安定固化法は,今後問題とされる各種使用済吸着剤の安定固化法の一つとして期待され,IRID委託研究として,日本原子力研究開発機構とユニオン昭和で2015年「汚染水処理二次廃棄物の圧縮固化及び焼結固化試験」,2016年「汚染水処理二次廃棄物の模擬物を用いた圧縮固化及び焼結固化試験」として実施しており,福島原発で発生している実際の廃棄物への適用技術としての検討が行われている。
2020年東京オリンピック開催に伴い,福島第一原子力発電所の汚染水処理問題が更にクローズアップされ,汚染水処理が加速化されている。現在,汚染地下水やトレンチ水の処理が進められており,廃炉過程で生じる汚染水処理等々の課題への取り組みも開始されている。また,現在懸案となっている不溶性フェロシアン化物スラッジ等,後処理に不安がある材料に対して,ゼオライトを利用した適切な安定化処理の方法も提案されており,ゼオライトを核としたこれらの吸着剤開発の取り組みが,被災地の復興,さらには地球環境保護の一助となれば幸いである。
謝辞Acknowledgments
本開発を行うにあたり,御支援頂きました三村均東北大学名誉教授(ユニオン昭和最高技術顧問),電力中央研究所小山研究参事,塚田副研究参事,土方上席研究員,東北大学多元研佐藤教授,桐島准教授,秋山助教,日本原子力研究開発機構山岸グループリーダーに深謝いたします。
引用文献References
1) 山岸 功,三村 均,出光一哉,福島第一原子力発電所高汚染水の処理処分の課題,日本原子力学会誌 特集記事,54, 166–170(2012).
2) 三村 均,山岸 功,秋葉健一,ゼオライトによる放射性セシウムとストロンチウムの除去,日本化学会誌,No. 3, 621~627(1989).
3) 松倉 実,安定固化可能なCs及びSr選択性吸着剤の実用化技術開発,日本吸着学会誌技術ハイライト,28, 12–16(2015).
4) 松倉 実,黒崎文雄,ゼオライトを主体とした高汚染水処理用吸着剤の開発,日本イオン交換学会誌,28, 51–57(2017).
5) H. Mimura, Zeolite, 29, 37(2012)。
6) M. Matsukura, Adsorption News, 28(4)(2015).
7) M. Matsukura, Ceramics, 52(1)(2017).
8) 土方孝敏,稲垣健太,塚田毅志,小山正史,石崎英司,松倉 実,日本原子力学会,福島第一原子力発電所放射性滞留水処理システムに関する研究開発(9).ゼオライトタイプ吸着剤のSr吸着速度定数の評価,秋期大会予稿集,N31(2013).
9) T. Hijikata, T. Tsukada, T. Koyama, E. Ishizaki, M. Matsukura, T. Kawada, H. Mizuno, Fall meeting of the Atomic Energy Society of Japan, G12(2014).
10) T. Asano, Y. Kani, Isotope News, 12, 716(2013).
11) H. Mimura, N. Fujita, H. Kanda, A. Kirishima, N. Sato and M. Matsukura, Proc.of Global 2015, paper 5015 September20–24, 2015, Paris, France.
12) H. Mimura, S. Susa, Y. Ito, Y. Saito, M. Matsukura, Proc.of 2ndInternational Conference on Nuclear Engineering, ICONE22, July7–14, 2014, PRAGUE, CZECH REPUBLIC.
13) Y. Ikarashi, Rana Syed Masud, H. Mimura, E. Ishizaki, M. Matsukura, Proc.of WM2013 Conference, Feb 24–28, 2013, Phoenix, Arizona, USA.
14) T. Nakai, S. Wakabayashi, H. Mimura, Y. Niibori, H. Tanikawa, E. Ishizaki, F. Kurosaki, M. Matsukura, Proc.of WM2013 Conference, Feb 24–28, 2013, Phoenix, Arizona USA.
15) H. Mimura, Y. Ikarashi, E. Ishizaki and M. Matsukura, Proc.of CIMTEC2014, 6th Forum on New Materials, June18, 2014, Montecatini Terme, Italy.
16) H. Mimura, S. Susa, Y. Ito, Y. Saito, M. Matsukura, Proc.of 22nd International Conference on Nuclear Engineering, ICONE22, July7–14, 2014, PRAGUE, CZECH REPUBLIC.