光学的に純粋な有機化合物は,医薬品,農薬,香料,液晶などの様々な製品に用いられている。光学活性化合物の需要が年々高まるなかで,環境に負荷をかけない合成法の開発も求められている。近年の金属触媒や有機分子触媒の発展によって,多種多様な不斉合成法が開発されてきたが,常温常圧でのエナンチオ選択性,官能基選択性,触媒回転率を考えると,酵素触媒反応を凌駕する反応は未だ少ない。さらに,発酵によって大量生産でき,かつ,生分解されて自然に帰すことができる酵素は,環境負荷が少なく,かつ,枯渇しない触媒である。一方で,天然の酵素が触媒する反応はかなり限定的である。様々な工夫によって酵素の利用を拡張することができれば,冒頭の要請に適う不斉合成法になる。
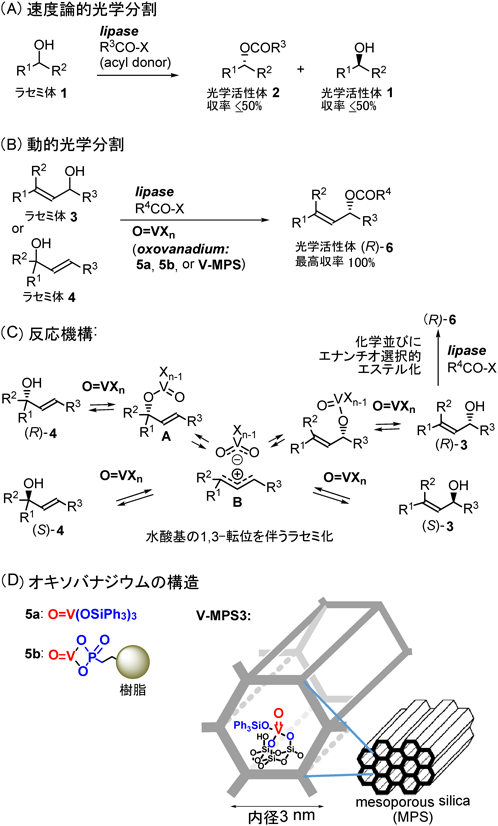
多様な酵素の中でも脂質のエステル結合を加水分解するリパーゼは,有機合成への利用件数が最も多い1)。それは,リパーゼは特に安定で,補酵素が要らず,かつ,非天然の幅広い基質に対して高い触媒活性,化学選択性,エナンチオ選択性を示すからである。さらに,リパーゼを有機溶媒中で用いると,カルボン酸とアルコールからエステルを形成する触媒にもなる。これまでリパーゼは,ラセミ体の速度論的光学分割(KR)に主に利用されてきたが,KRで得られる各エナンチオマーの収率は最大50%である(図1A)。もし,1つのフラスコ内で,KRとともに,KRで反応しなかったエナンチオマーのラセミ化を同時進行させることができると,ラセミ体を1つの光学活性体に収率100%で変換できる。本法は動的光学分割(dynamic kinetic resolution, DKR)と呼ばれ,この10余年で大きな進歩を遂げた2)。本稿では,DKRを達成するために著者らが独自に開発したバナジウム担持メソポーラスシリカ(V-MPS)の開発の経緯と応用について紹介する。
DKRには,通常,リパーゼと共にラセミ化触媒を用いる。DKRで良好な結果を得るためには幾つかの要件を満足する必要があるが (詳細は総説2)参照),そのなかで一番の難題は,リパーゼとラセミ化触媒が1つのフラスコ内で共存し,本来の機能を発揮できることである。と言うのは,リパーゼがもつ極性官能基がラセミ化触媒と反応して両触媒が失活しやすい,また,リパーゼが活性維持のために表面に抱えている多数の水分子によってラセミ化触媒が失活することもあるからである。現在,最も広く研究されている第二級アルコールのDKRでは,アルコールの酸化–還元反応によってラセミ化を触媒するルテニウム錯体が多用されている。ルテニウム錯体はリパーゼとの共存性に優れ,本DKR法は幅広い第二級アルコールに適用されてきたが2),アリルアルコールでは収率が低かった2a)。
3. 著者らの動的光学分割法:バナジウム担持メソポーラスシリカの開発の経緯
このような背景下,著者らは,オキソバナジウム種をラセミ化触媒として用い,アリルアルコール(3, 4)のDKR法の開発に着手した(図1B)。従来,バナジン酸エステルO=V(OR)3はアリルアルコールの水酸基1,3-転位を起こすことが知られていたが,150 °C以上の高温を要した3)。著者らは,この転位反応を再検討し,O=V(OSiPh3)3 5a4)または樹脂に結合したホスホン酸のオキソバナジウム(IV)化合物5b5)(図1D)が,アセトニトリル中,室温~50 °Cでアリルアルコール3の水酸基1,3-転位を起こすことを見出した。また,光学活性アルコール3では,ラセミ化も同時に起こることを発見した。種々の実験結果から,アルコールからバナジン酸エステルAが生じ,次いでC–O結合解裂によってイオン対Bが生成し,再結合の際にラセミ化が進行することがわかった(図1C)。
次に,ラセミ体3,5a (10 mol%)または5b (10 mol%),市販の固定化リパーゼ及び酢酸ビニルを反応させると,DKRが進行し,エステル(R)-6(光学純度90~99% ee)が収率64~99%で得られた(図1B)4,5)。これは,オキソバナジウムがラセミ化を伴いながら4種の異性体[(R)-3, (S)-3, (R)-4, (S)-4]間の動的平衡を生じ,リパーゼがその混合物のなかから(R)-3のみを高選択的にエステル化するという2つの反応が同時進行することで達成された(図1C)。リパーゼやオキソバナジウムを単独で用いては,この成果は得られない。また,本法では3の位置異性体4が等価な原料として利用できる(合成化学的応用例は後述)。しかし,5a,5bのラセミ化能は必ずしも高くなく,生成物の収率や光学純度が低いことがあった。また,ラセミ化能がより高いO=VSO4·nH2Oの利用を試みたが,リパーゼとの共存性が悪くDKRには適用できなかった。
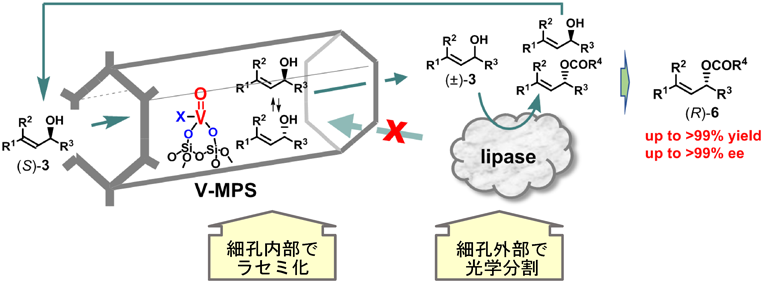
リパーゼとの共存性を維持しつつ,ラセミ化活性の高い触媒開発を目指して試行錯誤した結果,細孔径約3 nmのメソポーラスシリカMPS3[MPSの次の数字は細孔内径(単位nm)を示す]の細孔内表面にバナジウムを共有結合で固定した新規触媒V-MPS3を作成することを思い立った。すなわち,分子量数百以下の有機化合物はV-MPS3の細孔内に簡単に出入りすることができ,細孔内でラセミ化が進行する。一方,分子量数万ダルトン以上の巨大なリパーゼは細孔に入っていけず,細孔外で光学分割が進行する。こうして,オキソバナジウムによる『ラセミ化の反応場』と,リパーゼによる『光学分割の反応場』をMPSの細孔によって完全に分離するという考えである(図2)。なお,ゼオライト,モンモリロナイト,ハイドロタルサイトなどの多孔質または層状の無機化合物も同様に表面積が広いが,これらは酸性もしくは塩基性を示し,リパーゼに悪影響を及ぼす,もしくは,これら無機化合物による触媒作用でアシル化が進行する可能性があるので検討対象から除外した。
当時,著者は不勉強で多孔質無機素材のことを殆どわかっておらず,素人考えで,O=V(OSiPh3)35aのPh3SiO基がMPS3細孔内表面のシラノールに置換するのではないかと思い,5aとMPS3をベンゼン中で8時間,加熱環流した。生成物V-MPS3のICP-AMS分析から,バナジウム含量は0.20~0.22 mmol/gであった(同じ条件で,これまで十数回V-MPS3を合成したが,バナジウム含量の再現性は高い)。また,BET/BJH法で解析すると,MPSにバナジウムを固定したことで細孔内積,比表面積,細孔径が減少したことがわかった。また,元素分析結果などを総合して,V-MPS3は,細孔内表面の2つのシラノールによってオキソバナジウムが固定された構造(図1D)をとっていると推定している6a)。
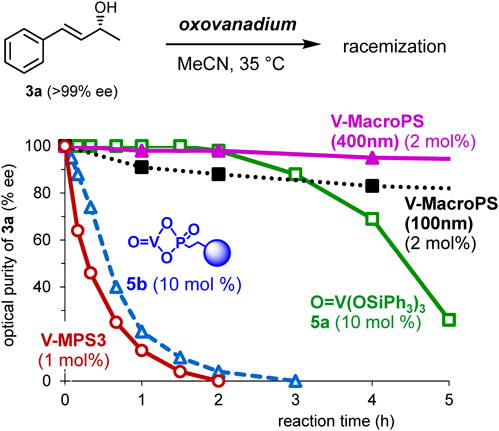
こうして調製したV-MPS3は,バナジウム量にしてわずか1 mol%で,従来使用していたラセミ化触媒5b (10 mol%)に匹敵する高いラセミ化活性を示した(図3)6a)。一方で,100 nmや400 nmの大きな細孔径を有するマクロポーラスシリカに結合したオキソバナジウム(V-MacroPS)はラセミ化活性が低いことから(図3),V-MPS3のナノサイズの細孔がラセミ化に極めて重要な役割を果たしていることが示唆された6a)。
また,V-MPS3を調製する際の加熱時間や温度によって,得られる触媒のラセミ化活性が変わることもわかった。例えば,ベンゼン中,長時間の加熱や,トルエン中加熱環流で得た触媒の活性は低かった。また,文献7)に従い合成したO=V(OSi)3MPS3型触媒(MPS3細孔内表面の3つのSi–OHで結合したオキソバナジウム)のラセミ化活性は低かった6a)。
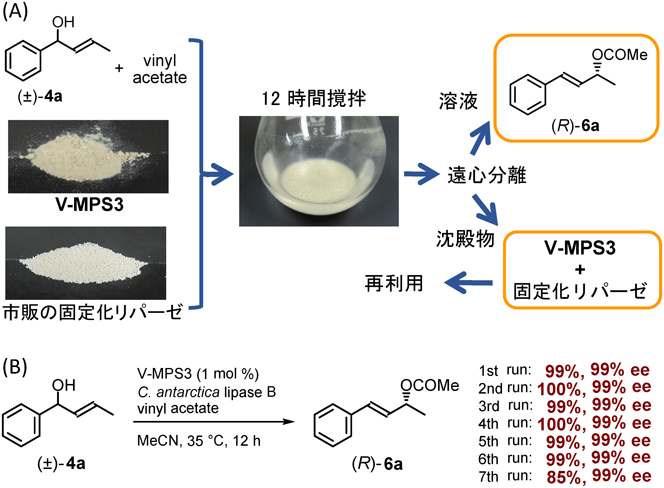
V-MPS3と市販のポリマー固定化リパーゼC. antarctica lipase B(CALB)はいずれも粉末である。基質アルコール(±)-4aをV-MPS (1 mol%),CALB,酢酸ビニルとともにアセトニトリル中35°Cで撹拌し,DKRを行った。反応後,溶液部分と沈殿物(V-MPS3とリパーゼの混合物)を分離した。溶液を減圧濃縮し,残渣を短いシリカゲルカラムに通すだけで高純度の6aが単離収率99%,光学純度99% eeで得られた(図4A)。また,溶液部分のICP分析の結果,バナジウムの漏洩量は0.0003%未満であった。さらに,沈殿物を減圧乾燥後,同じ条件で再度DKRに使用すると全く同じ結果が得られた。この混合触媒は6回再利用することができた(図4B)6a)。これらの結果は,V-MPS3とリパーゼの共存性が極めて高いことを示している。
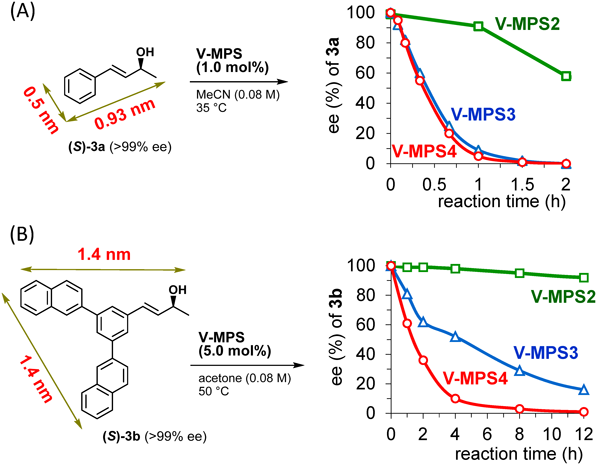
ところで,V-MPS3において,バナジウムはMPS3の外表面にも結合しているはずであるが,ラセミ化は本当にMPS3の細孔内部で進行しているのであろうか? また,通常,固体触媒は均一系触媒よりも触媒活性が低いのに対し,固体触媒であるV-MPS3が,なぜ,均一系触媒5aに比べて格段に高いラセミ化活性を有するのか?
これらの疑問に答えるため,V-MPS3に加えて,細孔径の大きさが2 nmと4 nmの2種のMPSにオキソバナジウムを結合したV-MPS2とV-MPS4を新たに作成した。V-MPS3を含めた3種のV-MPSを用いて,分子サイズの異なる光学活性アリルアルコールのラセミ化速度を比較した結果,分子サイズが1 nmよりも小さい3aのラセミ化速度はV-MPS2<V-MPS3=V-MPS4となった(図5A)。一方,分子サイズが最短1.4 nmの3bでは,V-MPS3とV-MPS4によるラセミ化速度にも顕著な違いが生じた(図5B)。このように,ラセミ化速度に関して,基質の分子サイズとV-MPSの細孔径には正の相関があることがわかった6b)。この事実は,ラセミ化は主に細孔内で進行することを強く示唆している。また,100 nmや400 nmの大きな細孔径を有するマクロポーラスシリカに結合したオキソバナジウム(V-MacroPS)のラセミ化活性が低いことから(図3),MPSの外表面に結合したバナジウムはラセミ化に殆ど寄与していないと考えられる。
では,なぜ,V-MPSの細孔内ではラセミ化が速いのか?
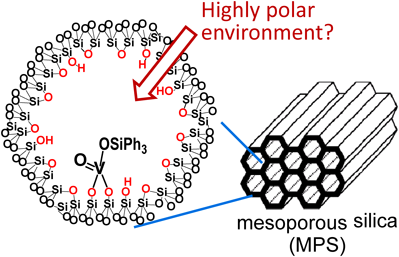
これまで5bやV-MPS3を用いたラセミ化実験で,ラセミ化は高極性溶媒の方が速いことがわかっている5,6a)。これは,極性環境が図1CにおけるAのC–O結合の解裂を促進し,また,イオン対Bを安定化するからと解釈できる。これらのことを踏まえて筆者らは,MPSの細孔内表面のSi–OHやSi–O–Si基による適度な極性空間(図6)8)がラセミ化を促進したのではないかと考察した。
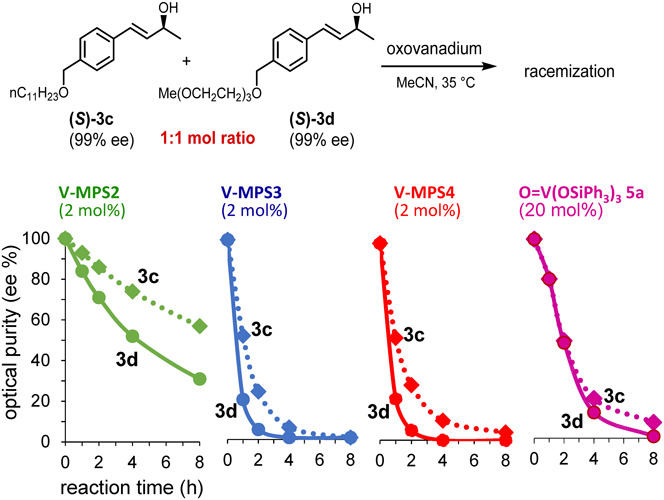
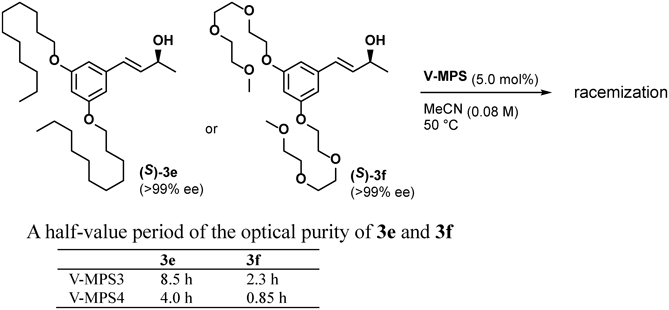
そこで,この仮説を検証すべく,長いアルキル鎖を持つ3c (>99% ee)と同じ長さのポリエーテル鎖を持つ3d (>99% ee)のモル比1 : 1混合物について,4種のオキソバナジウム[V-MPS2~4, O=V(OSiPh3)3 (5a)]を用いてラセミ化の経時変化を調べた(図7)。その結果,V-MPS2~4のいずれの場合も,3cより3dの方がラセミ化が速いことがわかった。一方,均一系触媒5a(V-MPS2~4の10倍のバナジウム量を用いた)によるラセミ化では3cと3dに殆ど差が無かった。これらの違いは,3cよりも極性が高い3dの方が細孔内に入りやすく,その結果,ラセミ化が速いことを物語っている。同様のラセミ化速度の違いは,2本のアルキル鎖を持つ3e (>99% ee)とポリエーテル鎖を持つ3f (>99% ee)の比較実験でも観察された(図8)。さらにこの実験では,V-MPS3とV-MPS4の活性の違いも顕著になった9)。
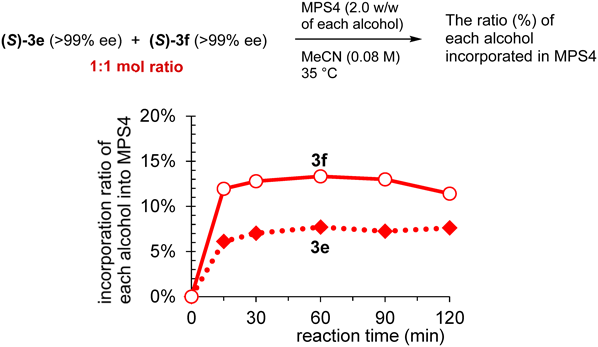
次に,3e (>99% ee)と3f (>99% ee)のモル比1:1混合物にMPS4を加え,細孔内への集積の割合を調べたところ,予想通り3eよりも3fの方が多く取り込まれることがわかった(図9)。なお,細孔内への移動は筆者らが予想した以上に速く,測定開始15分後には,ほぼ定常状態に達していた。
5. V-MPSとリパーゼを併用するDKRとその応用
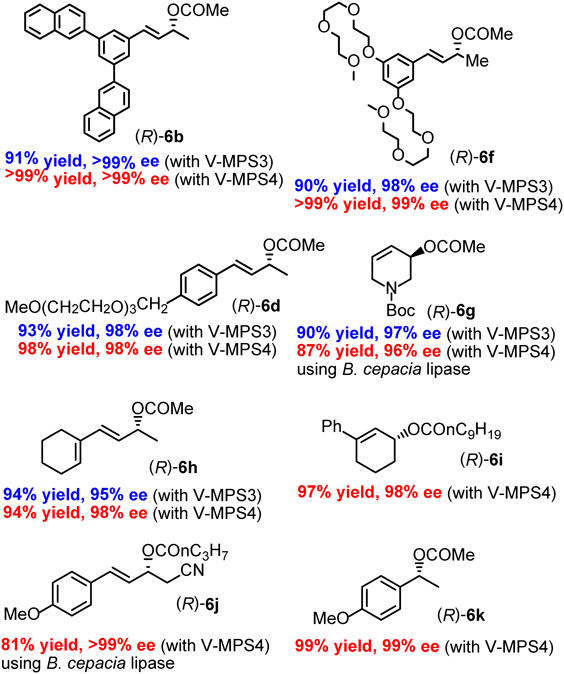
市販の固定化リパーゼとV-MPS3またはV-MPS4を同時に用いて,種々の第二級アルコールのDKRを行った。その結果の一部を図10に示す6b,10)。比較的大きなアルコールでは,前述のラセミ化速度の違いを反映して,V-MPS4の方がV-MPS3よりも対応するエステル[(R)-6b, (R)-6d, (R)-6f]をより高収率で与えた。一方,比較的小さなアルコールでは,V-MPS3とV-MPS4に殆ど差はなかった [(R)-6g,(R)-6h参照]。また,本法は環状アルコール,ベンジルアルコールなどにも適用できた[(R)-6i,(R)-6k参照]。このように,V-MPS4を用いれば,より広範囲の第二級アルコールのDKRを効果的に実施することができる11)。特筆すべき結果は,V-MPSは水酸基に反応してラセミ化を起こすが,生成する光学活性エステルとは反応しない,すなわち,生成物の光学純度を損なわないということである。その結果,多様なアルコールから光学純度98~99% eeのエステルが得られた。
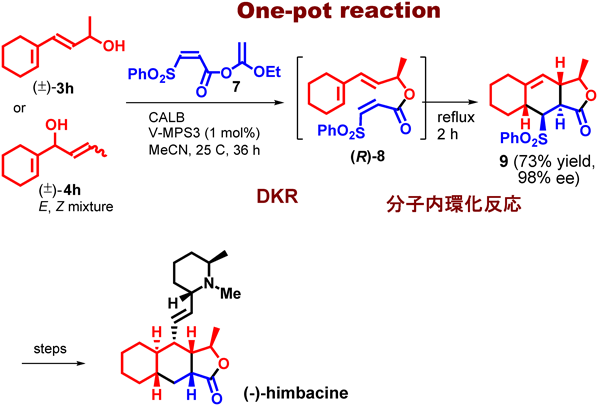
本DKR法の応用例を1つ挙げる。ジエン構造を含むラセミ体アルコール3hとジエノフィル構造を有するエステル7をCALB,V-MPS3と共に反応させると,DKRが進行して光学活性エステル8が生成した。8を単離することなく,反応液をそのまま加熱環流して分子内環化反応を起こし,3環性化合物9 (98% ee)を73%収率で得た。また,市販の反応剤から1工程で得られるラセミ体アルコール4h(E,Z混合物)からも同じ反応によって9 (98% ee)を同じ収率で得た。すなわち,(E)-4h,(Z)-4hともにV-MPSによる水酸基の1,3-転位を起こして3hに変換され,その後,DKRと分子内環化反応が進行したわけである。続いて,9を化学変換することによって(−)-himbacineの全合成を達成した(図11)12)。
このように,基質やアシル化剤を適切に設計することで,DKRによって導入されたアシル基を分子骨格構築に有効活用することができる(著者らは,アシル基を分子骨格構築に利用する類似の反応例を他にも報告している10,13))。また,アリルアルコールの水酸基の位置異性体(3hと4h)が等価な基質として利用できるために,合成ルートの設計の際に基質の選択肢が増えることも本DKR法の利点である。
リパーゼ(生体触媒)とオキソバナジウム化合物(金属触媒)という異質な触媒を1つのフラスコ内で同時に用いることにより,入手容易なラセミ体アルコールを光学的に純粋な化合物にほぼ定量的に変換する動的光学分割法を開発した。この際,高活性な2種類の触媒が互いに悪影響を及ぼすことなく共存し,本来の機能を最大限に発揮するためには,メソポーラスシリカのナノスケールの細孔を利用して両触媒を物理的に隔離することが極めて効果的であることもわかった。
また,この研究から,固体の細孔内部での反応にも関わらず,均一系触媒よりもラセミ化反応が速いこと(すなわち,細孔によってラセミ化が加速されること),極性化合物ほど細孔内に入りやすいことなど,細孔内の極性空間による反応特性を見出した。さらに,そのような反応性は内径数nmの狭隘な細孔内で発現し,細孔径100 nmでは発現しないことも明らかになった。また,そのような細孔内に基質アルコールが滞留することなく,迅速に出入りすることもわかった。
本文中にも述べたように,筆者らはMPSの細孔内にバナジウムを共有結合で固定するために多くの試行を重ねたが,最終的に筆者らが必要とした高い性能を持つV-MPS類を入手できたのは,幸運以外の何者でもない。なお,V-MPSによるラセミ化は陽イオン中間体を経て進行するため,基質の電子状態によってはラセミ化が極めて遅い場合もある。種々の酵素との共存性を確保しつつ,ラセミ化活性をさらに向上させることが目下の課題である。
最後に,多孔質無機素材の物性や反応性について未だ十分な検討も理解もできていない筆者が本稿を執筆したので,引用不足や間違った記述があれば,御教授頂けると幸いです。
謝辞Acknowledgments
本研究は共同研究者の熱心な実験と討論の賜物であり,深く感謝します。リパーゼは天野エンザイム株式会社,ロシュ・ダイアグノスティックス株式会社から,メソポーラスシリカは太陽化学株式会社から御恵与賜りました。V-MPSの分析に御助言や御指導いただいた佐治木弘尚教授(岐阜薬科大学),坂田昌弘教授(静岡県立大学),光延聖博士(同),ICP分析を行ってくださいましたエヌ・イーケムキャット株式会社,和光純薬工業株式会社に感謝します。また,本研究は文部科学省科研費[JP24106738(集積反応化学),JP26670002,JP15H04631,JP16H01151(中分子戦略)],日本医療研究開発機構創薬等ライフサイエンス研究支援基盤事業助成金によって行うことができました。関係者に深く感謝します。
引用文献References
1) 総説・成書:(a) 廣瀬芳彦,有合化誌,69, 506 (2011). (b) K. Faber, “Biotransformations in Organic Chemistry; A Textbook”, 6th Ed., Springer: Heidelberg, 2011. (c) “Future Directions in Biocatalysis”, 2nd Ed., Ed. by T. Matsuda, Elsevier: Amsterdam, 2017.
2) 総説:(a) S. Akai, Chem. Lett., 43, 746 (2014). (b) S. Takizawa, H. Gröger, H. Sasai, Chem. Eur. J., 21, 8992 (2015). (c) O. Verho, J.-E. Bäckvall, J. Am. Chem. Soc., 137, 3996 (2015). (d) O. Långvik, T. Saloranta, D. Y. Murzin, R. Leino, ChemCatChem, 7, 4004 (2015). (e) S. de Miranda Amanda, S. M. Miranda Leandro, O. M. A. de Souza Rodrigo, Biotechnol. Adv., 33, 372 (2015). (f) 赤井周司,有合化誌,75, 441 (2017).
3) (a) P. Chabardes, E. Kuntz, J. Varagnat, Tetrahedron, 33, 1775 (1977). (b) S. Bellemin-Laponnaz, J. P. Le Ny, A. Dedieu, Chem. Eur. J., 5, 57 (1999). (c) S. Bellemin-Laponnaz, J.-P. Le Ny, Compt. Rend. Chim., 5, 217 (2002).
4) S. Akai, K. Tanimoto, Y. Kanao, M. Egi, T. Yanamoto, Y. Kita, Angew. Chem. Int. Ed., 45, 2592 (2006).
5) S. Akai, R. Hanada, N. Fujiwara, Y. Kita, M. Egi, Org. Lett., 12, 4900 (2010).
6) (a) M. Egi, K. Sugiyama, M. Saneto, R. Hanada, K. Kato, S. Akai, Angew. Chem. Int. Ed., 52, 3654 (2013). (b) K. Sugiyama, Y. Oki, S. Kawanishi, K. Kato, T. Ikawa, M. Egi, S. Akai, Catal. Sci. Technol., 6, 5023 (2016).
7) G. Du, S. Lim, M. Pinault, C. Wang, F. Fang, L. Pfefferle, G. L. Haller, J. Catal., 253, 74 (2008).
8) メソポーラス化合物の親水性と細孔表面構造に関する論文:(a) E.-P. Ng, S. Mintova, Microporous Mesoporous Mater., 114, 1 (2008). (b) S. Maaz, M. Rose, R. Palkovits, Microporous Mesoporous Mater., 220, 183 (2016).
9) メソポーラスシリカの細孔サイズによる選択的反応の例:(a) M. Iwamoto, Y. Tanaka, N. Sawamura, S. Namba, J. Am. Chem. Soc., 125, 13032 (2003). (b) T. Shinoda, Y. Izumi, M. Onaka, J. Chem. Soc. Chem. Commun., 1801 (1995).
10) S. Kawanishi, K. Sugiyama, Y. Oki, T. Ikawa, S. Akai, Green Chem., 19, 411 (2017).
11) 現在,V-MPS4は和光純薬工業株式会社から販売されている.
12) K. Sugiyama, S. Kawanishi, Y. Oki, M. Kamiya, R. Hanada, M. Egi, S. Akai, Bioorg. Med. Chem. (2017), in press [DOI:10.1016/j.bmc.2017.08.019].
13) (a) S. Akai, T. Naka, S. Omura, K. Tanimoto, M. Imanishi, Y. Takebe, M. Matsugi, Y. Kita, Chem. Eur. J., 8, 4255 (2002). (b) S. Akai, K. Tanimoto, Y. Kita, Angew. Chem. Int. Ed., 43, 1407 (2004). (c) H. Nemoto, K. Tanimoto, Y. Kanao, S. Omura, Y. Kita, S. Akai, Tetrahedron, 68, 7295 (2012).