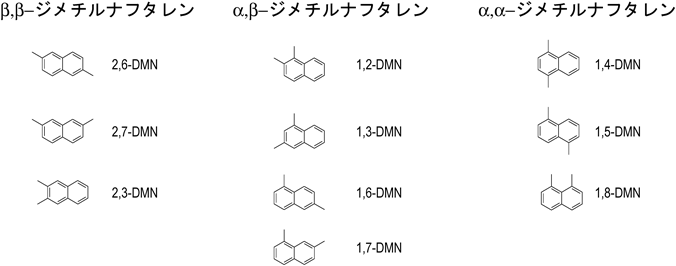
ポリエチレンナフタレート(PEN)は,ヤング率,引っ張り強度,ガス透過性などのほとんどの特性においてポリエチレンテレフタレート(PET)に比べ優れたエンジニアリングポリマーであり,エチレングリコールと2,6-ジメチルナフタレン(2,6-DMN)から得られる2,6-ナフタレンジカルボン酸より合成される。一方,2,6-ジメチルナフタレンの製造には多段階の反応が用いられるとともに,多量の溶媒が用いられる1)。さらに,ジメチルナフタレンは10種の異性体[β,β-DMN(2,6-DMN, 2,7-DMN, 2,3-DMN), α,β-DMN(1,2-DMN, 1,3-DMN, 1,6-DMN, 1,7-DMN), α,α-DMN(1,4-DMN, 1,5-DMN, 1,8-DMN)](図1)を有し,これらの沸点が同程度であることから複雑な分離工程が必須となる2,3)。ゼオライトは炭化水素の分子径に近い細孔径を有することからMFI型,EUO型,FAU型ゼオライト,モルデナイトなどを用いた2-メチルナフタレン(2-MN)のメチル化による2,6-DMN選択合成が検討されてきた4–8)。MFI型ゼオライトはナフタレンの最小分子径に比べ細孔径が小さく,細孔内における原料/生成物の拡散抵抗が大きいため,β,β-DMNの選択率が高い。また,MFI型ゼオライト構造中のAlをFeで同型置換したフェリシリケートを用いることで,さらにβ,β-DMN選択率が向上することが報告されている9)。一方で,上述の理由から原料/生成物のゼオライトの細孔内での拡散抵抗が非常に大きいため,転化率の向上が難しい。これに対し細孔径の大きいゼオライトを用いることで原料/生成物の拡散係数の向上を図れるが,ベータやY型ゼオライトのように大きい細孔を有するゼオライトでは拡散抵抗の低減から高い転化率を得られるものの,生成物の異性化反応や逐次的なメチル化反応によって目的生成物の選択率が低下する。このため,高2-MN転化率,高β,β-DMN選択率の両立可能なゼオライト触媒の設計が求められる。
MTW型ゼオライトは酸素12員環1次元細孔構造を有するゼオライトであり,その細孔径はβ,β-DMNの分子径と同程度であることから,β,β-DMN合成に有効な触媒であると期待される。また,ゼオライト触媒を用いた反応では,その見かけの反応速度はゼオライトの細孔内での原料/生成物の拡散速度と活性点上での反応速度の関係に影響を受けるため,反応工学の観点からに触媒を設計する上で反応速度の情報が必要となる。しかし,気相での2-MNのメチル化反応ではMTW型ゼオライトに限らず,逐次的なメチル化により生成する高分子量の炭素質やコークが細孔入口付近に析出,滞留することで反応中に触媒の活性を著しく低下させる。このため,コーク析出の影響が無視小の反応速度の情報を得ることは容易ではない。筆者らは,本反応に対しアルキル化剤でもあるメタノールの超臨界流体を反応場に用いることで,反応中のゼオライト触媒上へのコーク析出を気相での反応に比べ劇的に抑制できることを明らかにするとともに,2-MNのメチル化反応の反応速度解析を行ってきた。本稿では,MTW型ゼオライトの粒子径,酸量,Feによる同型置換が2-MNメチル化反応に及ぼす影響について反応工学の観点から検討した最近の研究について紹介する。
MTW型ゼオライトは一般的な水熱合成法を用いて合成した。Si源にコロイダルシリカ,Al源にアルミン酸ナトリウム,Fe源に硝酸鉄九水和物,アルカリ源に水酸化ナトリウムを用いた。有機構造規定剤(OSDA)にはメチルトリエチルアンモニウムクロリド(MTEACl)あるいはテトラエチルアンモニウムブロミド(TEABr)を用いた。Si源,Al源あるいはFe源,アルカリ源を含む水溶液を調製した後,本溶液をオートクレーブに移し,423 K, 6日間,水熱合成を行うことで白色固体を得た。得られた白色固体は洗浄,乾燥の後,空気中823 Kで焼成した。得られた試料の構造解析にはX線回折装置(Ultima IV,リガク),細孔容積およびBET表面積の解析には77 Kにおける窒素吸着測定装置(Belsorp mini,マイクロトラック・ベル)を用いた。また,試料の結晶形態は走査型電子顕微鏡(JSM-6500F,日本電子)を用いて観察した。
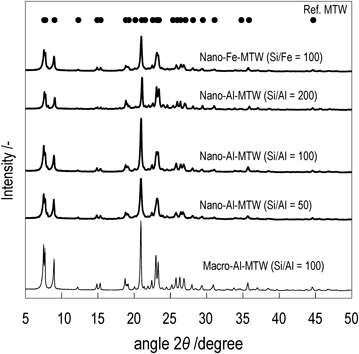
図2は合成した試料のXRDパターンを示しており,いずれの試料においてもMTW型ゼオライトに起因するピークを示した。図3に得られたMTW型ゼオライトのFE-SEM像を示す。Al-MTW(Si/Al=100)は,異なる有機構造規定剤を用いることで一次粒子径が1000~4000 nmのMTW型ゼオライト[Macro-Al-MTW(Si/Al=100)]と,50~100 nmの一次粒子が凝集し700 nm程度の二次粒子を形成したMTW型ゼオライト [Nano-Al-MTW(Si/Al=100)] が得られた。また,XRDパターンからシェラー式により算出した粒子径はそれぞれ2940 nm, 76 nmであった。Al量の異なるMTW型ゼオライト [Al-MTW(Si/Al=50, 200)]およびフェリシリケート[Fe-MTW(Si/Fe=100)]についても50~100 nmの一次粒子が凝集し二次粒子を形成していることが確認された。表1は窒素吸着測定およびac-NH3-TPD測定10)により得られた特性評価の結果であり,得られたゼオライトは既報のMTW型ゼオライトが有するミクロ孔容積およびBET表面積と同程度の値であった11,12)。Nano-Fe-MTW(Si/Fe=100)のUV-visスペクトルはFe種がクラスターや凝集状態でなく単独で存在すると考えられる300 nm以下でのみ吸収スペクトルを示した。さらに,nano-Fe-MTW(Si/Fe=100)はnano-Al-MTW(Si/Al=100)とほぼ等しい酸量であったことから,Feはゼオライト骨格中に存在していると考えらえる。
表1. 合成した試料の細孔特性,Si/T(T=Al or Fe),酸量.| Sample | Vm/cm3 g−1 | SBET/m2 g−1 | SEXT/m2 g−1 | Si/T ratio/- | Acid site amouunt/mol kg−1 |
|---|
| Macro-Al-MTW(Si/Al=100) | 0.13 | 340 | 8 | 104 | 0.15 |
| Nano-Al-MTW(Si/Al=50) | 0.13 | 340 | 40 | 56 | 0.28 |
| Nano-Al-MTW(Si/Al=100) | 0.13 | 310 | 45 | 107 | 0.15 |
| Nano-Al-MTW(Si/Al=200) | 0.11 | 290 | 30 | 233 | 0.07 |
| Nano-Fe-MTW(Si/Fe=100) | 0.13 | 300 | 40 | 102 | 0.15 |
| Vm: micropore volume, SBET: BET surface area(BET method), SEXT: external surface area(t-plot method), Si/T ratio is measured by XRF. |
2-MNのメチル化反応は回分式反応器を用いて行った。硝酸アンモニウム水溶液を用いてイオン交換後,823K,1時間焼成した触媒0.5 gと2-MNとメタノールの混合溶液(2-MN/CH3OH=0.1(物質量比))を反応容器に入れ,反応温度(523~583 K)まで昇温した後,90~270分反応を行った。反応圧力は,触媒量と混合溶液の重量比を固定し,反応器に入れる総量を変化させることで制御した。反応後の混合液は濾過により触媒と生成液に分離し,生成液をGC-FID(GC-2010,島津製作所)により分析した。また,反応後の触媒上に生成したコークは熱重量分析計(TGA-50,島津製作所)を用いて測定した。
3.1. 反応圧力の影響
ゼオライト触媒の活性低下要因の一つに反応中のコーク析出が挙げられる。多環芳香族化合物のアルキル化やアルキル基の成長,環化によってコークは成長するため14),空間制約のないゼオライト外表面上にコークが析出,成長することで細孔閉塞が生じ,反応活性が低下する。森本らは超臨界水中に重質炭化水素が分散しやすく,重質炭化水素の分解生成物間の反応による高分子量化,コーク析出の抑制が可能であることを報告している15)。そこで,コーク析出の抑制を目的としてnano-Al-MTW(Si/Al=100)を用い,反応圧力2.9および25 MPaで2-MNのメチル化反応を573 Kで行った。メタノールの臨界温度,臨界圧力はそれぞれ512.6 K,8.1 MPaであることから,2.9 MPaでは気相,25 MPaでは超臨界流体中での反応と考えられる。表2に2-MNの転化率と生成物の選択率を示す。反応圧力が高くなることにより2-MN転化率は大きくなり,α,α-DMNの生成は抑制された。さらに,反応圧力2.9,25 MPaにおけるコーク析出量はそれぞれ13, 0.5 wt%であり,超臨界流体中で反応を行うことによりコーク析出が大幅に抑制された。メタノールに対する有機物の溶解度は圧力の増加に伴い向上することが知られている16)。気相反応では,2-MNのメチル化反応により生成したβ,β-DMNの揮発性は低く,触媒からの脱離が起こりにくいために触媒上での滞留時間が長くなり,α,α-DMNへの異性化や逐次反応の進行によるコーク析出が促進されたと考えられる。一方,有機物の溶解性が高い超臨界流体中で反応を行うことにより,生成物がバルク流体中に速やかに移動し,上述の逐次的な反応が抑制されることにより,コーク析出の抑制が達成されたと考えられる。
表2. 2-メチルナフタレンの転化率および生成物の選択率に対する反応圧力の影響(反応温度:573 K,反応時間:90分).| Pressure/MPa | Conversion/mol% | Selectivity/mol% | DMN isomer fraction/mol% | Coke loading/wt% |
|---|
| DMN | 1-MN | β,β-DMN | α, β-DMN | α,α-DMN |
|---|
| 2.9 | 3.9 | 22.5 | 77.5 | 42.5 | 48.8 | 8.7 | 13.1 |
| 25 | 6.6 | 21.3 | 78.7 | 45.1 | 54.9 | 0 | 0.5 |
3.2. 1-MNのメチル化反応
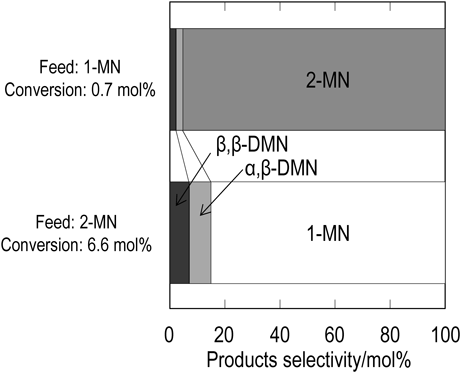
MTW型ゼオライトを用いた2-MNのメチル化反応では,DMNの生成と同時に2-MNの異性体である1-MNが生成する。そのため,2-MNのメチル化反応で生成する1-MNのメチル化反応によるDMN生成が考えられる。図4は,反応温度573 K,反応圧力25 MPa,反応時間90分の条件でnano-Al-MTW(Si/Al=100)を用いて1-MNのメチル化反応を行った時の1-MNの転化率と生成物の選択率を示す。比較のため,同一反応条件で行った2-MNのメチル化反応の結果をあわせて示す。1-MNの転化率は0.7 mol%と2-MNの転化率である6.6 mol%に比べ低い転化率を示した。また,1-MNのメチル化反応の主たる生成物は1-MNの異性体である2-MNであり,DMNの選択率は非常に小さい値であった。Milliniらは,MTW型ゼオライトの細孔内において1-MNが受ける拡散抵抗は2-MNが受ける拡散抵抗の5倍以上であることを報告している17)。1-MNはMTW型ゼオライトの細孔内を拡散しづらいため,MTW型ゼオライトの外表面上で1-MNが異性化することで2-MNが生成したと考えられる。また,α位にメチル基を有するDMNは,1-MNに比べMTW型ゼオライトの細孔内での拡散抵抗が大きいことから,α,β-DMNはゼオライトの外表面上酸点でβ,β-DMNの異性化により生成したと考えられる。さらに,1-MNのメチル化によるα,α-DMN生成が確認されないことから,ゼオライト外表面上でのメチル化反応は本反応条件では無視小であると推測される。そこで以降では,超臨界メタノール中における2-MNのメチル化反応は2-MNのメチル化によるDMN生成と2-MNの異性化による1-MN生成の並列反応として反応速度解析を行った。
3.3. 結晶サイズの異なるMTW型ゼオライトを用いた2-MNのメチル化反応の速度解析
反応容器内には2-MNに対しアルキル化剤であるメタノールが大過剰に存在するため,DMNおよび1-MNの生成は2-MN濃度に対して一次で反応が進行すると仮定することができる。よって,メチル化反応,異性化反応の反応速度式はそれぞれ(1)式,(2)式で表すことができる。
ここで,rm, riはそれぞれメチル化,異性化の反応速度(mol kg−1 s−1),km,kiはそれぞれメチル化,異性化の反応速度定数(m3 kg−1 s−1),Cは2-MN濃度(mol m−3)を表す。
(1)式,(2)式より
ここで,Wは触媒重量(kg),Vは反応物体積(m3)である。
(3)式を積分することで(4)式が得られる。
ここで,C0は2-MNの初期濃度(mol m−3),tは反応時間(s)をそれぞれ表す。
(4)式は,2-MNのメチル化および異性化反応が2-MN濃度に対し一次で進行する場合,ln(C0/C)値が時間に対して線形で表されることを示している。また,(1)式および(3)式から(5)式が得られる。
ここで,CDMN,0,CDMNはそれぞれDMNの初期および反応時間tにおける濃度(mol m−3)を示している。
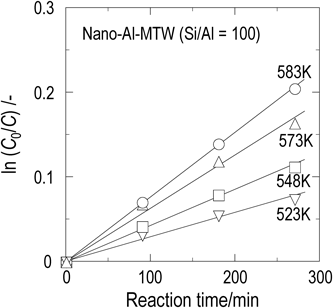
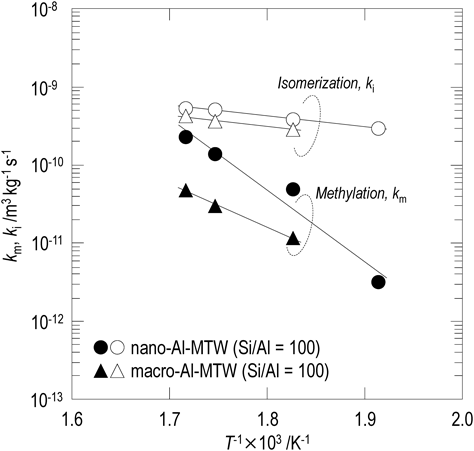
ゼオライト触媒を用いた反応では,一般的に活性点上での反応速度とゼオライトの細孔内における原料/生成物の拡散速度の関係が見かけの反応速度に影響する。ゼオライトの細孔内を拡散する分子の拡散速度は,(有効拡散係数)/(拡散距離)2で表すことができ,拡散距離を短くすることで拡散速度の向上が図れる。つまり,ゼオライトの粒子径を小さくすることで拡散抵抗を低減できることが明らかになっている18)。そこで,酸量が等しく粒子径の異なるMTW型ゼオライトを用いて2-MNのメチル化反応を行った。表3に,粒子径の異なるMTW型ゼオライト(Si/Al=100)を用い,反応温度583 K,反応圧力25 MPa,反応時間270分の条件で2-MNのメチル化反応を行った時の2-MNの転化率,各生成物への選択率,DMN異性体の生成物分布を示す。粒子径を小さくすることで,2-MNの転化率およびDMNへの選択率が向上した。次に,523~583 K,反応時間0~270分の条件で2-MNのメチル化反応を実施し,ln(C0/C)値の経時変化を得た。一例としてnano-Al-MTW(Si/Al=100)の結果を図5に示す。反応温度に依らず,得られたプロットは原点を通る直線で相関された。このことは,上記で仮定したように2-MNのメチル化反応,異性化反応が2-MN濃度に対して一次で進行することを示している。また,図5の直線の傾きから各温度におけるメチル化反応,異性化反応の速度定数の和を得た。さらに,反応初期および反応時間tにおける2-MNとDMNの各濃度を用い,(5)式から各反応温度における(km+ki)/ki値を算出した。これらの連立方程式を解くことで,メチル化反応の速度定数(km)および異性化反応の速度定数(ki)を求めた。図6に粒子径の異なるMTW型ゼオライト(Si/Al=100)を用いて得られた2-MNのメチル化反応および異性化反応の反応速度定数のアレニウスプロットを示す。Nano-Al-MTW(Si/Al=100)を用いた際の異性化反応の反応速度定数は,macro-Al-MTW(Si/Al=100)を用いた際の異性化反応の反応速度定数に比べ高い値を示した。一方,nano-Al-MTW(Si/Al=100)の外表面積はmacro-Al-MTW(Si/Al=100)の5倍程度大きいにも関わらず,nano-Al-MTW(Si/Al=100)とmacro-Al-MTW(Si/Al=100)の異性化反応の反応速度定数の比は1.2~1.4であり,外表面積の比に比べ小さい値であった。Derouaneらは,ZSM-5の粒子径によりZSM-5の外表面とバルクとでAl分布が異なることを報告している19)。そこで,粒子径の異なるMTW型ゼオライトを用い,MTW型ゼオライトの細孔径よりも大きい分子径を有する1,3,5-トリイソプロピルベンゼン(TIPB)の接触分解反応20)を行った。本反応は固定層流通式反応器を用い,反応温度573 Kで行った。MTW型ゼオライト外表面上でのTIPBの接触分解反応は,TIPB濃度に対し一次で進行するとして速度解析を行った。Macro-Al-MTW(Si/Al=100)およびnano-Al-MTW(Si/Al=100)のTIPBの接触分解反応の反応速度定数はそれぞれ3.8 m3 kg−1 s−1,8.1 m3 kg−1 s−1と求まり,nano-Al-MTW(Si/Al=100)のTIPB接触分解反応の反応速度定数はmacro-Al-MTW(Si/Al=100)に比べて約2倍大きいことがわかった。これは,2-MNの異性化反応の反応速度定数の比に近い値であり,MTW型ゼオライトにおいても粒子径が異なることでAlの分布が異なり異性化の反応速度に影響を及ぼすことを示唆している。一方,異性化反応の反応速度定数より求められる活性化エネルギーは粒子径に依らず約26 kJ mol−1とほぼ等しい値を示した。
表3. 2-メチルナフタレン転化率および生成物の選択率に対するAl-MTW粒子径の影響(反応温度:573 K, 反応圧力:25 MPa,反応時間:270分).| Sample | Conversion/mol% | Selectivity/mol% | DMN isomer fraction/mol% | Coke loading/wt% |
|---|
| DMN | 1-MN | β,β-DMN | α, β-DMN | α,α-DMN |
|---|
| Macro-Al-MTW(Si/Al=100) | 10.2 | 6.6 | 93.4 | 45.5 | 54.5 | 0 | 0.8 |
| Nano-Al-MTW(Si/Al=100) | 15.1 | 21.4 | 78.6 | 44.9 | 55.1 | 0 | 1.0 |
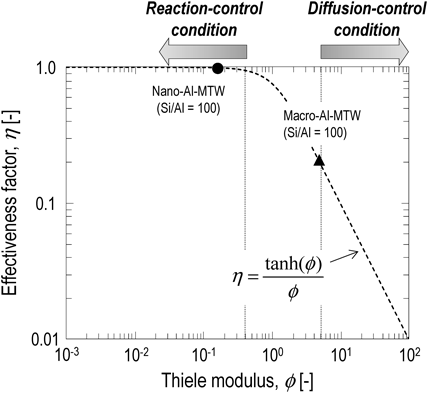
Nano-Al-MTW(Si/Al=100)の2-MNメチル化反応の反応速度定数は,macro-Al-MTW(Si/Al=100)に比べ高い値を示した。また,nano-Al-MTW(Si/Al=100)の2-MNメチル化反応の反応速度定数より求められる活性化エネルギーは176 kJ mol−1であり,macro-Al-MTW(Si/Al=100)の105 kJ mol−1に比べ高い値を示した。拡散律速下で反応が進行する際,得られる反応速度定数から求められる活性化エネルギー(Eobs)は,真の活性化エネルギー(反応律速下で反応が進行する際に得られる反応速度定数から求められる活性化エネルギー,E)の半分の値(Eobs=E/2)となることが知られている21)。Macro-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応の反応速度定数より求められる活性化エネルギーは,nano-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応の反応速度定数から求められる活性化エネルギーの半分の値に近い値であった。よって,macro-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応は,触媒粒子内における2-MN, DMNの拡散の影響を強く受けて反応が進行しており, nano-Al-MTW(Si/Al=100)を用いたメチル化反応の反応速度定数に比べて低い反応速度定数が得られたと考えられる。さらに,粒子径の異なるmacro-Al-MTW(Si/Al=100),nano-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応の反応速度定数(反応温度:583 K)を用い,各触媒の触媒有効係数(η)を粒径変化法22,23)により算出した。触媒有効係数は,「見かけの反応速度」と「ゼオライトの細孔内を拡散する分子の拡散抵抗の影響が無視小である時の反応速度(真の反応速度)」の比であり,反応律速下で反応が進行している場合には触媒有効係数は1に近い値をとり,拡散律速下で反応が進行している場合には触媒有効係数は0.2より小さい値をとる24)。図7にmacro-Al-MTW(Si/Al=100)とnano-Al-MTW(Si/Al=100)の触媒有効係数(反応温度:583 K)を示す。Nano-Al-MTW(Si/Al=100)の触媒有効係数は0.99であり,ほぼ1に近いことからnano-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応(反応温度:583 K)は反応律速下で反応が進行していることが確認された。一方,粒径変化法により求めたmacro-Al-MTW(Si/Al=100)を用いたメチル化反応の触媒有効係数は0.21であり拡散律速に極めて近い条件で反応が進行していると考えられる。本解析からも,macro-Al-MTW(Si/Al=100)を用いた2-MNのメチル化反応の反応速度は,MTW型ゼオライトの細孔内における2-MN,DMNの拡散の影響を強く受けており,主たる反応場は触媒外表面に近い領域のみとなるため, 反応速度定数はnano-Al-MTW(Si/Al=100)を用いたメチル化反応に比べ低い値が得られたと考えられる。Foglerは,触媒有効係数が0.95以上で触媒粒子内における分子の拡散が得られる反応速度に及ぼす影響が無視小になるとしている21)。そこで,触媒有効係数が0.95となるときのAl-MTW(Si/Al=100)を求めたところ,240 nmよりも小さいAl-MTW(Si/Al=100)を用いることでMTW型ゼオライトの細孔内における2-MN,DMNの拡散の影響が無視小な反応律速下で2-MNのメチル化反応(583 K)を進行させることが可能であることが明らかになった。
3.4. MTW型ゼオライトの酸量,Feによる同型置換の影響25)
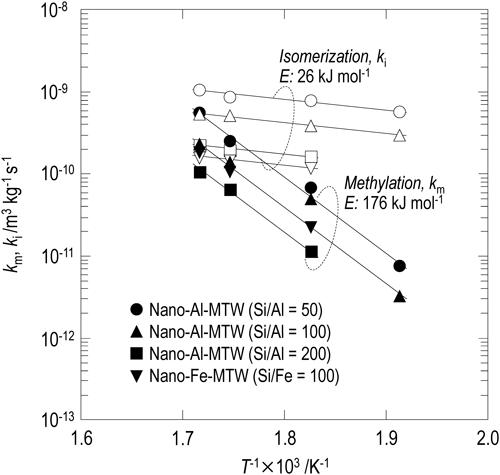
MTW型ゼオライトの酸量および骨格の原子種が2-MNのメチル化,異性化反応の反応速度に及ぼす影響を検討するため,nano-Al-MTW(Si/Al=50, 100, 200)およびnano-Fe-MTW(Si/Fe=100)を用いて同様に反応速度解析を行った。図8に2-MNのメチル化および異性化反応の反応速度定数のアレニウスプロットを示す。Nano-Al-MTWのSi/Al比を変化させたところ,骨格中のAl量の増加(酸量の増加)に伴い2-MNのメチル化反応および異性化反応の反応速度定数は大きくなった。これは酸量の増加に伴い2-MNのメチル化反応,異性化反応の活性点が増加したためと考えられる。また,2-MNのメチル化反応,異性化反応の反応速度定数より求められる活性化エネルギーはMTW型ゼオライトのSi/Al比に依らずそれぞれ176 kJ mol−1,26 kJ mol−1であった。上述のように,拡散律速下で反応が進行する場合,得られる反応速度定数から求められる活性化エネルギーは真の活性化エネルギーよりも低い値となる。2-MNのメチル化反応の反応速度定数から求められる活性化エネルギーはSi/Al比に依らず176 kJ mol−1であることから,nano-Al-MTWのSi/Al比によらず反応律速下で2-MNのメチル化反応が進行していると考えられる。
Nano-Fe-MTW(Si/Fe=100)を用い,同様に2-MNのメチル化,異性化反応の速度解析を行った。2-MNのメチル化反応の反応速度定数はnano-Al-MTW(Si/Al=100)を触媒に用いた反応で得られる反応速度定数とほぼ等しい値であった。一方,異性化反応の反応速度定数は低下した。これは,反応律速下でAl-MTWと酸量の等しいFe-MTWを2-MNのメチル化反応に用いることで,Al-MTWで得られる2-MNのメチル化反応の反応速度を維持したまま,副反応である2-MNの異性化反応を抑制していることを示している。
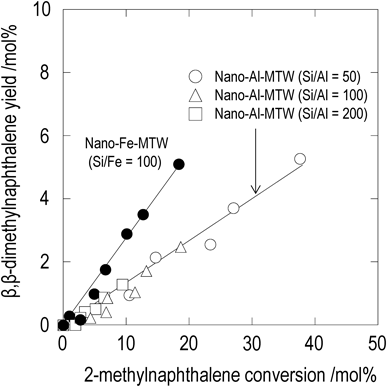
図9に2-MNの転化率とβ,β-DMNの収率の関係を示す。2-MNの転化率とβ,β-DMNの収率はFe-MTW,Al-MTWそれぞれに対して1本の直線で相関された。これは,転化率が同一である場合,β,β-DMNの収率がSi/Al比や反応温度に依らずほぼ等しいことを示している。一方,Fe-MTWはAl-MTWに比べ同一の2-MN転化率において高いβ,β-DMN収率を示した。Fe-MTWを用いることで,2-MNの異性化反応による1-MN生成と同様に, β,β-DMN(2-MNのメチル化反応の生成物)の異性化反応によるα,β-DMN生成が抑制されたためと考えられる。
引用文献References
1) N. Kraikul, P. Rangsunvigit, S. Kulprathipanja, Chem. Eng. J., 114, 73(2005).
2) K. Bobuatong, M. Probst, J. Limtrakul, J. Phys. Chem. C, 114, 21611(2010).
3) R. Millini, C. Perego, Top. Catal., 52, 42(2009).
4) S. B. Pu, T. Inui, Appl. Catal. A, 146, 305(1996).
5) H. Klein, H. Fuessa, S. Ernst, J. Weitkamp, Microporous Mater., 3, 291(1994).
6) J. N. Park, J. Wang, S. I. Hong, C. W. Lee, Appl. Catal. A: Gen., 292, 68(2005).
7) D. Frankel, M. Cherniavsky, B. Ittah, M. Levy, J. Catal., 101, 273(1986).
8) J. Weitkamp, M. Neuber, Stud. Surf. Sci. Catal., 60, 291 (1991).
9) T. Komatsu, Y. Araki, S. Namba, T. Yashima, Stud. Surf. Sci. Catal., 84, 1821(1994).
10) T. Masuda, Y. Fujikata, S. R. Mukai, K. Hashimoto, Appl. Catal. A: Gen., 165, 57(1997).
11) B. Gil, L. Mokrzycki, B. Sulikowski, Z. Olejniczak, S. Walas, Catal. Today, 152, 24(2010).
12) X. Wei, P. Panagiotis, G. Smirniotis, Micropor. Mesopor. Mater., 89, 170(2006).
13) G. Watanabe, Y. Nakasaka, T. Taniguchi, T. Yoshikawa, T. Tago, T. Masuda, Chem. Eng. J., 312, 288(2017).
14) L. Pinard, S. Hamieh, C. Canaff, F. F. Madeira, I. Batonneau-Gener, S. Maury, O. Delpoux, K.B. Tayeb, Y. Pouilloux, H. Vezin, J. Catal., 299, 284(2013).
15) M. Morimoto, Y. Sugimoto, Y. Saotome, S. Sato, T. Takanohashi, J. Supercrit. Fluids, 55, 223(2010).
16) F. Hassan, B. A. Duri, J. Wood, Chem. Eng. J., 207–208, 133(2012).
17) R. Millini, F. Frigerio, G. Bellussi, C. Perego, P. Pollesel, U. Romano, J. Catal., 217, 298(2003).
18) H. Konno, R. Ohnaka, J. Nishimura, T. Tago, Y. Nakasaka, T. Masuda, Catal. Sci. Technol., 4, 4265(2014).
19) E. G. Derouane, J. P. Gilson, Z. Gabelica, C. M. Desbuquoit, J. Verbist, J. Catal., 71, 447(1981).
20) S. Inagaki, S. Shinoka, Y. Kaneko, K. Takeuchi, R. Komatsu, Y. Tsuboi, H.Yamazaki, J.N. Kondo, Y. Kubota, ACS. Catal., 3, 74(2013).
21) H. S. Fogler, Elements of Chemical Reaction Engineering, 4th Ed., Prentice Hall,(2013).
22) W. O. Haag, R. M. Lago, P. B. Weisz, Faraday Discuss. Chem. Soc., 72, 317(1982).
23) A. Shichi, K. Katagi, A. Satsuma, T. Hattori, Appl. Catal. B: Environ., 24, 97(2000).
24) O. Levenspiel, Chemical Reaction Engineering, 2nd Ed., John Wiley & Sons,(1972).
25) G. Watanabe, Y. Nakasaka, T. Masuda, J. Jpn. Petrol. Inst., 60, 146(2017).