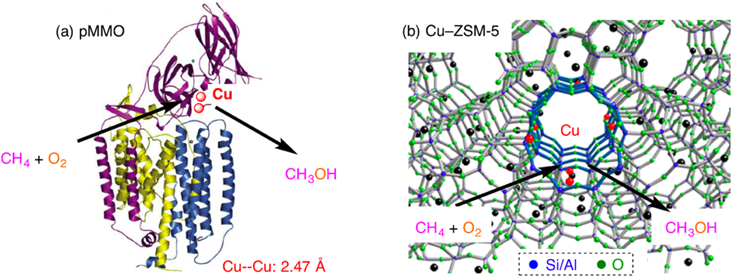
メタンは最も不活性な炭化水素である。これは,メタンの大きなHOMO–LUMOギャップ,双極子モーメントの欠如,さらに,高いC–H結合の結合エネルギー(104 kcal/mol)に由来する。この不活性な炭化水素は,我が国近海にメタンハイドレートの形で存在するため,その有効利用が望まれている。この有効利用の一例が,メタンから工業的に重要なメタノールに変換することである。現在,メタンからメタノールへの変換プロセスは,合成ガス(一酸化炭素と水素ガス)を経由した二段階反応からなる。この二段階反応は熱力学的に不利なプロセスである。一方,メタン直接酸化によるメタノール転換は熱力学的に有利であるため,メタンの直接酸化を効率よく行う触媒の開発が望まれる。このメタン直接酸化を触媒する材料を開発する上で,生体酵素であるメタンモノオキゲナーゼ(MMO(図1a))1)の触媒機能に注目が集まっている。
MMOは,その内部に含まれる配位環境の不飽和な鉄や銅元素1)を利用して,メタンと酸素を常温常圧下でメタノールに変換する。メタンが最も不活性な炭化水素であることを考えると,MMOの触媒機能は驚異的である。この驚異的な機能を模倣した触媒の開発に向けて数多くの研究が行われているが,ここでは不飽和な配位環境を有する遷移金属元素を安定に存在させることが重要になる。これを実現するため,金属錯体を合成するときに複雑な配位子を用いたり,ナノメートルサイズのゼオライト細孔を用いたりする(図1b)2)。この生体酵素模倣触媒のひとつとして銅担持ゼオライトが挙げられる。本稿では,銅担持ゼオライト触媒の開発に向けた実験報告に触れ,それに関連した筆者の理論計算の結果を紹介する。
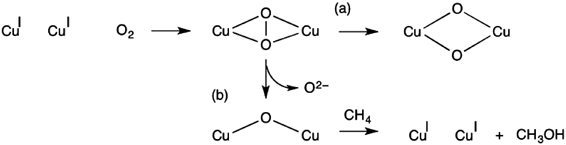
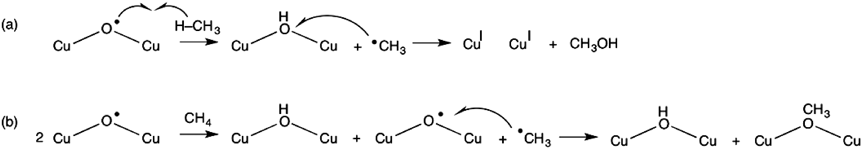
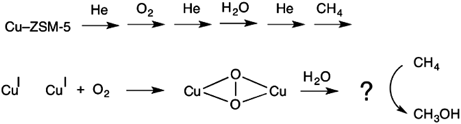
メタン直接酸化を行う銅担持ゼオライト触媒の開発における先駆的な研究は,2005年SelsやSchoonheydtらによって発表された2)。この研究では,酸素雰囲気下の銅担持ゼオライト(Cu‒ZSM-5)が,メタンをメタノールに変換することを見出している(図1b)。この反応はMMOのメタン酸化反応に類似したものであり(図1a),生体酵素からの類推により銅二核サイトが反応に関与していると予測されている2)。実際,銅二核サイトと酸素分子が結合することにより反応が開始し,その後,酸素分子は活性化され活性種に至る。特に,22700 cm−1に光吸収ピークを有する活性種は,200°Cでメタンをメタノールに変換する2)。このメタン直接酸化反応の詳細な機構を調べるため,銅二核サイトと酸素分子が反応することにより生じる中間体の同定が行われている。2005年に発表された論文では,MMOの場合の類推からスキーム1aのように反応が進行するものと予想されている2)。
このスキームでは,銅二核サイトに酸素分子が結合し酸素結合種を生成した後,銅カチオンから電子が移動することにより酸素分子が活性化される。その結果,酸素結合種はbis(μ-oxo)種に変形する。MMOの場合,このbis(μ-oxo)種がメタンを直接酸化しメタノールに変換する活性種であり,銅担持ゼオライトの場合もこの種がメタンを活性化するものと考えられていた。その後,2009年SelsらはSolomonらのグループとともに共鳴ラマン分光法測定を行い,22700 cm−1に光吸収ピークを有する活性種が456および870 cm−1にピークを有することを明らかにした3)。このラマンピークは酸素同位体効果を有し,この実験事実を説明することのできる種としてCu‒O‒Cu構造が提案されている(スキーム1b)。
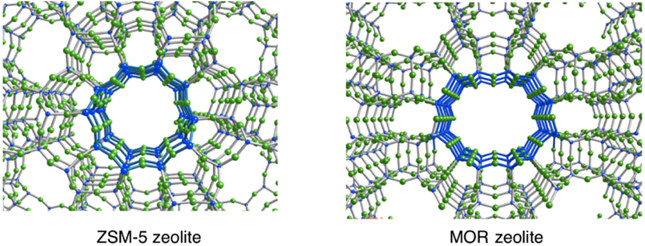
酸素雰囲気下銅担持ゼオライトのメタン活性化能は,ZSM-5型でのみならず,モルデナイト(MOR)型4,5)やSSZ-13型6)においても発現する。特に,酸素雰囲気下銅担持MORゼオライトにおけるメタン–メタノール変換反応はvan Bokhovenらによって精力的に研究されている4,5)。この銅担持MORゼオライトと酸素分子との反応で生じる活性種も,ZSM-5ゼオライトの場合と同様に22700 cm−1付近に光吸収ピークを有する。従って,銅担持MORゼオライトの場合もCu‒O‒Cu種が生成することが予想される。しかし,MORゼオライトは12員環細孔を有するため,10員環細孔を有するZSM-5型ゼオライトとは触媒活性を有するものと考えられる(図2)。実際,銅担持MORゼオライトによるメタン–メタノール転換反応における活性化エネルギー(11.1および14.7 kcal/mol)4,5)は,銅担持ZSM-5ゼオライトの場合(15.7 kcal/mol)2)よりも低い。
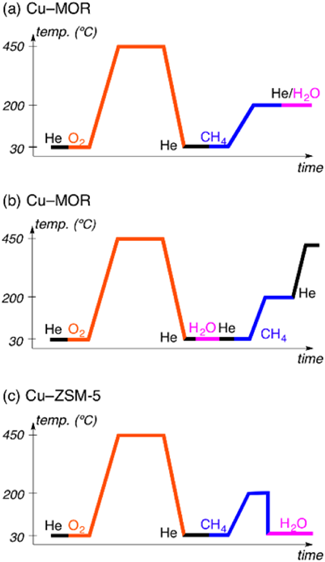
ここで,酸素雰囲気下銅担持MORゼオライトにおけるメタン–メタノール変換反応のプロセスの詳細を紹介する(図3a)4,5)。このプロセスでは,酸素分子が銅担持MORゼオライトに挿入された後,450°Cで加熱処理を行う。この加熱処理では,Cu‒O‒Cu種が生成するものと考えられている。その後,室温まで下げた活性種とメタンを200°Cで反応させる。この時,メタンのC‒H結合は橋架け酸素により活性化され,メチルラジカルと水酸基が生じる(スキーム2)。その後の経路として二つが考えられる。一つ目では,メチルラジカルと水酸基が再結合してメタノールが生成する。これがメチルラジカルリバウンド機構である(スキーム2a)。もう一つでは,生成したメチルラジカルが他のCu‒O‒Cu種に結合し,メトキシ基と水酸基が別々に生成する(スキーム2b)。その後,メトキシ基からメタノールを生成するために水分子を挿入する(図3a)4,5)。この水分子の挿入のタイミングは,銅担持MORゼオライトのメタンの活性化能に影響を与える。実際,Cu‒O‒Cu種の生成後,水分子を挿入しメタンと反応させた場合(図3b),メタノールの生成量が図3aのプロセスとは異なっていた。したがって,酸素雰囲気下の銅担持MORゼオライトにおけるメタン–メタノール変換反応において水分子の存在が重要になることが予想される。銅担持ZSM-5ゼオライトの場合は,図3aのプロセスと同様にメタン酸化反応が行われている(図3c)2)。
上述の実験結果を理解するうえで,ゼオライト細孔内部で生じる活性種の構造を“原子レベル”で理解する必要がある。これを行う最も有効な手段の一つが第一原理密度汎関数法計算である。密度汎関数理論により,全系の基底状態のエネルギーが電子密度ρの汎関数として一意的に決まることが保証され,さらに一電子密度を決める式が与えられる。実際には,コーン・シャム(Kohn–Sham, (KS))方程式を解くことに帰着される。この際,KS方程式の演算子の計算に用いた初期の電子密度が,方程式を解くことにより得られる電子密度と一致するまで計算を続ける(自己無撞着場の方法)。その後得られる全エネルギーEは,電子密度ρとコーン・シャム軌道φKSを用いて以下のように記述される。
右辺の第一項は一つの電子の運動エネルギーの和,第二項は核引力エネルギーの和の平均,第三項は古典的なクーロン反発ポテンシャルである。第四項Excは交換相関項と呼ばれ,Hartree–Fock法などの相互作用のない参照電子系では記述できない多電子効果を表している。現在,交換相関項を表すために様々な汎関数が開発されており,ある問題を解決するための最適な汎関数を選択することが計算精度を大きく左右する。
式(1)に示すように密度汎関数法計算で得られる全エネルギーには電子相関を直接取り込んでいるため,ある程度精密な計算が可能である。さらに他の電子相関を考慮した計算方法にくらべ計算コストが低いため,数百原子からなる大規模系に適用可能である。このため,大規模密度汎関数法計算で得られる情報は,実験データと比較可能である。例えば,ある最適化構造における原子間結合距離はX線構造解析のデータと比較できるし,波動関数のエネルギー準位や波動関数の形(特に,軌道の対称性)は,物質の光吸収発光を理解するうえで不可欠な情報である。また最適化構造において振動解析(エネルギーの二次微分の計算)を行うことで,ラマン分光で得られる振動スペクトルとの対比が可能である。さらに,量子化学計算ではエネルギー的に不安定な局所構造や遷移状態も得ることができる。遷移状態からは化学反応の進みやすさの指標である活性化エネルギーがわかり,反応設計には不可欠な情報である。
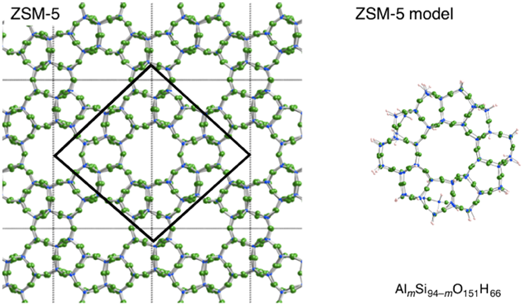
筆者らは,酸素雰囲気下の銅担持ZSM-5ゼオライト(Cu‒ZSM-5)での活性種の構造,およびそのC‒H結合の活性化機構を,密度汎関数法計算を用いて明らかにしている。ZSM-5のナノメートルサイズの10員環細孔を表現したモデルとしてSi94O151H66を用いた(図4, 左実線部分)。このモデルの10員環細孔のいくつかのSi原子をAl原子に置換し,ZSM-5ゼオライトを作成した。その後,このAlmSi94−mO151H66モデルに遷移金属元素を挿入した。このモデルは,ZSM-5そのものの10員環細孔を保持しているため,7,8)Cu‒ZSM-5の酸素分子活性化で生成する反応中間体の構造,およびそのC‒H結合の活性化機構を議論することが可能である。
4. 銅担持ZSM-5ゼオライト触媒に関する理論的研究
4.1. 銅担持ZSM-5ゼオライトの酸素分子活性化で生じる反応中間体の構造特性9)
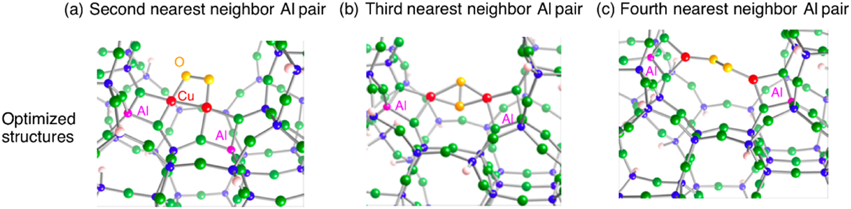
銅担持ZSM-5ゼオライトの酸素分子活性化においてどのような反応中間体(酸素種)が生成するかを密度汎関数法計算で調べた9)。ここで,電子相関を表すためにB3LYP汎関数を用いた。上述の実験報告に従い,これらの酸素種は,銅二核サイトが酸素分子との反応により生じると考えた。ZSM-5ゼオライト内部の銅二核サイトにおいて,その銅原子間距離は二つのアルミニウム原子の配置に依存して様々な値(2.39~6.31 Å)をとる9)。この結果は,銅二核サイトを形成する時,アルミニウム原子の分布が重要になることを示唆している。このため,MMOの場合(銅原子間距離;2.47 Å)と比較して,ゼオライト内部の銅二核サイトと酸素分子との結合種(酸素結合種)は様々な構造をとる可能性がある。実際,図5から,アルミニウム原子配置に依存して酸素結合種の結合様式が異なることがわかる。実際,二つのアルミニウム原子が近い場合,シス型エンドオン酸素結合種が生成するものの,二つのアルミニウム原子が遠い場合はトランス型エンドオン酸素結合種となる。一方,アルミニウム原子間隔がその中間の場合,酸素分子はサイドオン型で二核銅カチオンに配位する。この時,銅カチオンと酸素分子との相互作用により酸素分子は活性化されるが,その活性化の度合いは酸素結合種の構造に依存した。
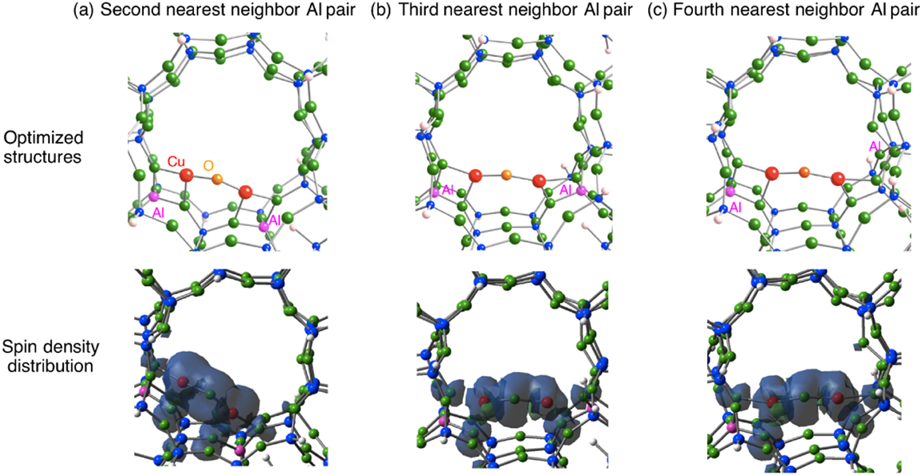
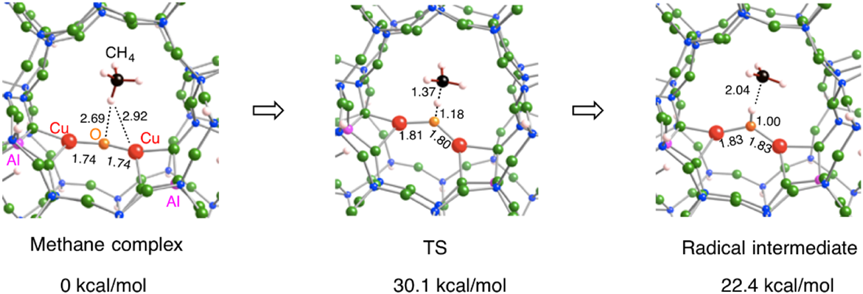
次に,酸素結合種を450°Cの加熱によって生じると考えられるCu‒O‒Cu種の構造特性について触れる。ゼオライト内部Cu‒O‒Cu種の構造最適化においても,先ほどの三つのアルミニウム原子配置を考えた。スピン多重度として,不対電子を持たない一重項と,不対電子を二つ持つ三重項を考え構造最適化を行った(図6)。その結果,すべての配置において,三重項状態のほうが一重項状態よりエネルギー的に安定であった。この三重項状態のスピン密度分布から,橋架け酸素原子に顕著なスピン密度が存在することがわかった。この結果は,橋架け酸素原子がラジカル性を有していることを示している。このため,このラジカル酸素原子を利用することによりメタンのC‒H結合はホモリティックに解裂し,メチルラジカルと橋架け水酸基が生成されるものと予想される。実際,橋架け酸素原子によるメタンのC‒H結合の解裂の遷移状態を求めることによって,そのポテンシャルエネルギー曲面の情報を得た(図7)。ここで遷移状態探索には,Quadratic Synchronous Transit(QST)法を用いた。図7から,橋架け酸素原子によるメタンのC‒H結合の解裂の活性化エネルギーは30 kcal/molであることがわかる。この値は,幾分実験値(15.7 kcal/mol)2)よりも高い値であった。一方,MMOの場合の類推から予想されたbis(μ-oxo)種についても同様の調査を行った結果,スピン一重項状態のほうが三重項状態よりもエネルギー的に安定であった。この構造では,アルミニウム原子配置に依存して銅カチオンの配位環境が大きく異なり,その結果,構造の安定性に影響を与えることがわかった。この結果は,bis(μ-oxo)種のゼオライトの制限された空間への閉じ込めが反応中間体の安定性を支配することを示唆している9)。
4.2. 酸素雰囲気下銅担持ZSM-5ゼオライトのメタン直接酸化反応における水分子の影響10)
酸素雰囲気下銅担持ZSM-5ゼオライトの関連する実験において,メタンのC‒H結合の活性化を司るCu‒O‒Cu種の生成には450°Cの加熱が必要であった。この高温加熱は,常温常圧でメタンを活性化するMMOを模倣した触媒を開発するうえで大きな障害になる。この課題を解決するヒントを得るため,筆者らはスキーム3のようなプロセスを考え,その反応機構に関する知見を密度汎関数法計算により得た。このプロセスでは,銅二核サイトと酸素分子が結合して酸素結合種が生成した後,水分子を挿入するものである。この時,酸素結合種は水分子と何らかの反応を起こし活性な種に変形し,その後,メタンをメタノールに変換するものと予測される。このプロセスは,酸素分子活性化に必要な熱エネルギーが不要であるという利点を有している。
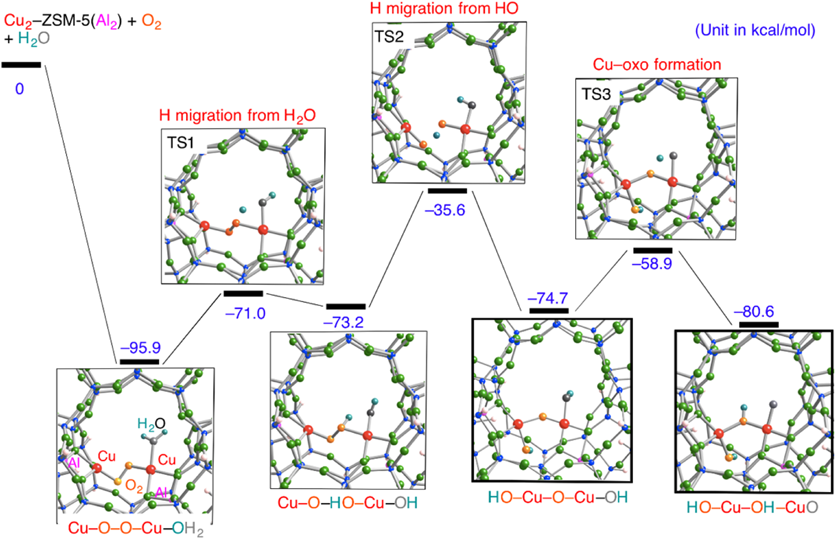
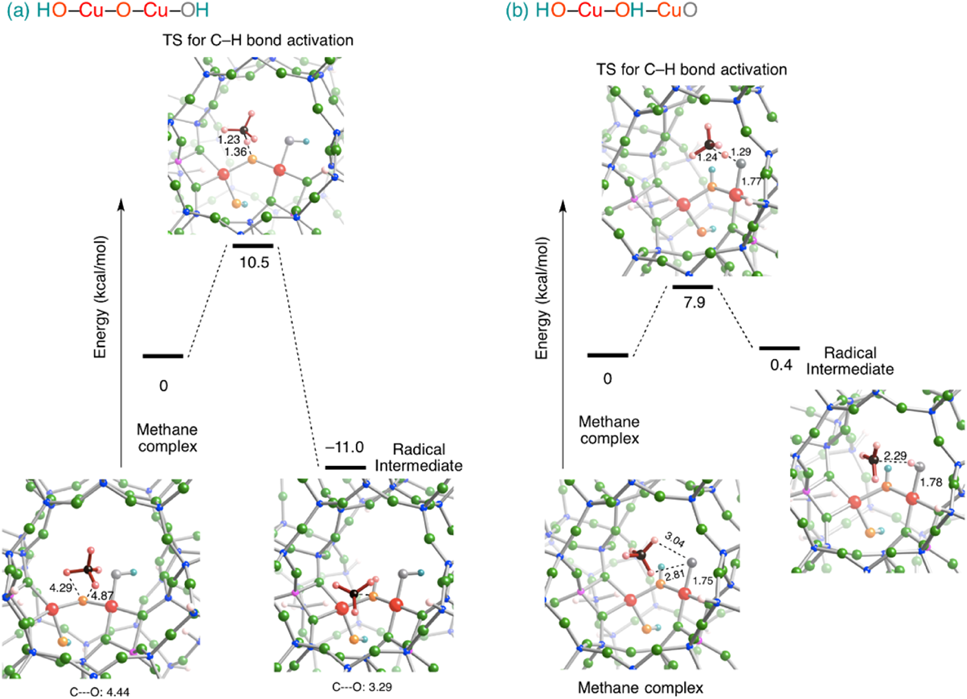
図8に,水1分子存在下Cu‒ZSM-5における酸素分子活性化反応における反応中間体および遷移状態を示す。この反応プロセスは酸素活性化Cu‒ZSM-5に水分子が結合することで始まる(図8)。次に,水分子の水素原子が酸素原子に移り水酸基が新たに生成する。この水酸基の水素原子はもう一方の酸素原子に移り,さらにこれと同時にO–O結合が開裂することによってHO‒Cu‒O‒Cu‒OH種に至る。その後,水酸基の水素原子が酸素原子に転移しHO‒Cu‒OH‒CuO種が生成する。このプロセスにおけるポテンシャルエネルギー曲面に注目したところ,すべての反応中間体および遷移状態は反応の初段階よりもエネルギー的に安定である。このため,水一分子存在下Cu‒ZSM-5における酸素分子活性化反応は自発的に進行するものと考えられる。
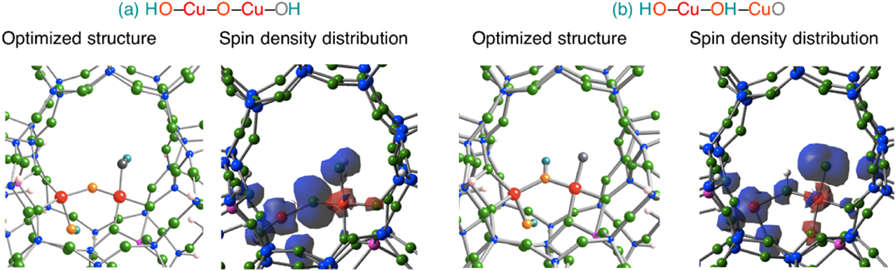
ここで,このプロセスで生じるHO‒Cu‒O‒Cu‒OH種およびHO‒Cu‒OH‒CuO種の特性を調べるため,それぞれのスピン密度分布を図9に示した。図9より,HO‒Cu‒O‒Cu‒OH種では橋架け酸素原子に,HO‒Cu‒OH‒CuO種では終端酸素原子にそれぞれスピン密度が局在することがわかる。このことは,この二つの反応中間体にラジカル酸素原子が存在することを意味する。これらのスピン密度分布から判断すると,HO‒Cu‒O‒Cu‒OH種はCu‒O‒Cu種と近い特性を有するものと考えられる。一方,ラジカル酸素原子を端に有するHO‒Cu‒OH‒CuO種は,気相中で生成するCuO+種に類似するものと考えられる。11)このCuO+種は,Schwarzらのグループによって作成され,メタンに高い化学活性を有することが報告されている。11)したがって,水一分子存在下Cu‒ZSM-5における酸素分子活性化反応で生じる反応中間体はメタンのC‒H結合を効率よく活性化する可能性がある。
この予測に従い,HO‒Cu‒O‒Cu‒OH種およびHO‒Cu‒OH‒CuO種によるC‒H結合の活性化機構を調べた(図10)。いずれの中間体においてもラジカル酸素原子を有するため,メタンのC‒H結合はホモリティックに解裂し,メチルラジカルと水酸基が生成するものと考えられる。実際,密度汎関数法計算で得られた反応機構もスキーム2に従うものであった。HO‒Cu‒O‒Cu‒OH種の場合,橋架け酸素原子によりメタンのC‒H結合を解裂するには10.5 kcal/molのエネルギーが必要であり,HO‒Cu‒OH‒CuO種の終端酸素原子によるC‒H結合の解裂には7.9 kcal/molのエネルギーが必要であった。ここで,Cu‒O‒Cu種,HO‒Cu‒O‒Cu‒OH種,およびHO‒Cu‒OH‒CuO種のラジカル酸素原子によるメタンのC‒H結合解裂に必要な活性化エネルギーを比較する。図7および10より,Cu‒ZSM-5の酸素分子活性化反応において水一分子が存在した場合に生じる反応中間体のほうが,水が存在しない場合に比べて活性化エネルギーが低いことがわかった。この結果から,銅二核サイトと酸素分子が結合した後,水一分子を挿入することにより,メタンへの反応性が高い化学種の生成が示唆される。この水一分子の挿入は,銅二核サイトの酸素分子活性化を促進する効果も有しているため,このプロセスは,水分子が挿入されない場合よりもエネルギー的に有利である。したがって,上述の密度汎関数法計算で得られた結果は,常温常圧でメタンを活性化するMMOを模倣した触媒を開発するうえで有用な情報を与えるものと考えられる。
謝辞Acknowledgments
本研究を行うにあたり有益な知見を与えて下さった京都工芸繊維大学小林久芳教授および岡山大学黒田泰重教授に厚く御礼申し上げる。また,本研究は文部科学省科学研究費補助金「新学術領域研究」感応性化学種が拓く新物質科学,および若手研究(B)から財政的支援を頂いた。
引用文献References
1) R. L. Lieberman and A. C. Rosenzweig, Nature, 434, 177–182 (2005).
2) M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 127, 1394–1395 (2005).
3) J. S. Woertink, P. S. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt, and E. I. Solomon, Proc. Natl. Acad. Sci., 106, 18909–18913 (2009).
4) E. M. Alayon, M. Nachtegaal, M. Ranocchiari and van J. A. Bokhoven, Chem. Commun., 48, 404–406 (2012).
5) E. M. Alayon, M. Nachtegaal, A. Bodi and van J. A. Bokhoven, ACS Catal., 4, 16–22 (2014).
6) M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Chem. Commun., 51, 4447–4450 (2015).
7) T. Yumura, T. Nanba, H. Torigoe, Y. Kuroda and H. Kobayashi, Inorg. Chem., 50, 6533–6542 (2011).
8) T. Yumura, T. Kumondai, T. Wakasugi, Y. Kuroda and H. Kobayashi, RSC. Adv., 7, 4950–4959 (2017).
9) T. Yumura, M. Takeuchi, H. Kobayashi and Y. Kuroda, Inorg. Chem., 48, 508–517 (2009).
10) T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, ACS Catal., 6, 2487–2495 (2016).
11) N. Dietl, C. van der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Angew. Chem. Int. Ed., 50, 4966–4969 (2011).