結晶性Mo3VOx複合酸化物(MoVO)は {Mo6O21}6− 5員環ユニットと{MO6} (M=Mo, V)金属酸素八面体が複雑に配列した板(a–b面)がc軸方向に積層して形成する棒状の結晶である1–3)。a–b面ではこれらのユニットの縮合により構造内に5,6,および7員環が形成し,このうち7員環は直径0.40 nmのミクロ細孔として機能する4)。7員環細孔径は酸化還元により制御可能である5–6)。これはレドックス能に優れたMo,Vを構成元素とすることに由来し,レドックス能の低いSiやAlで構成されるゼオライトや有機物をリンカーとするMOFには見られない興味深い現象である。また,MoVOはアルコール,アルデヒド,低級アルカンの選択酸化反応について,その洗練された触媒構造を反映した極めて高い触媒活性を示す7–11)。このような性質から,MoVOは高い酸化活性を示す八面体モレキュラーシーブとして大きく注目されている。
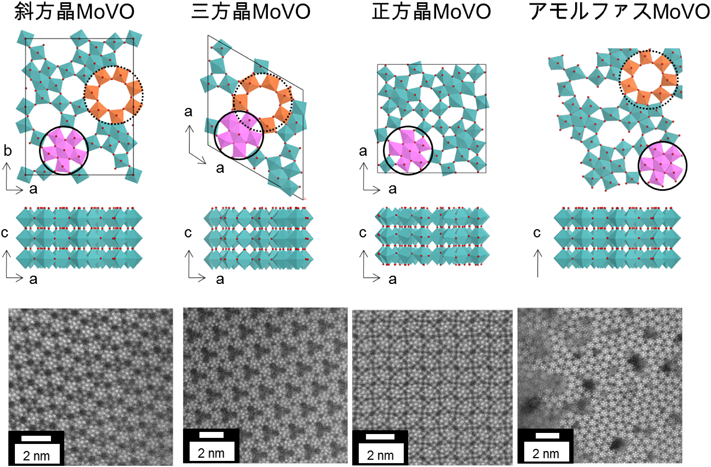
さらに近年では{Mo6O21}6− 5員環ユニットと{MO6} (M=Mo,V)金属酸素八面体で構成される同様の組成の棒状結晶で,断面部の構造のみが異なる斜方晶MoVO,三方晶MoVO,正方晶MoVO,および積層方向の配列は規則的であるが断面部がディスオーダーであるアモルファスMoVOの作りわけに成功した10–12)(Fig. 1)。これらの結晶相の比較から,触媒反応における結晶面の強い依存性が見て取れた。さらに斜方晶MoVOについてはエタン選択酸化反応をモデル反応として,その触媒構造と触媒機能に強い関係を見出すことができた13–16)。本稿ではMoVOの結晶形成プロセスから触媒機能評価に至るまで,最新の展開も含めて概説する。
MoVOは(NH4)6Mo7O24·4H2O(AHM)とVOSO4·5.4H2Oの混合溶液を水熱合成することで得られる。この合成において,透明なAHM溶液と青色のVOSO4溶液を混ぜ合わせると深い紫色の溶液が得られる。この溶液を溶液Ramanや溶液UVにより検討したところ,溶液内には[Mo72V30O282(H2O)56(SO4)12]36−({Mo72V30})が形成しており,{Mo72V30}中のMo6+とV4+間の原子価間電荷移動により紫色に呈色していることが分かった11),16–19)。前駆体溶液のpHを変えて水熱合成を行ったところ,MoVOの形成はpHに敏感であった。Table 1に溶液UVにより測定した種々pHにおけるMoVO前駆体溶液中の{Mo72V30}量と形成した結晶の結晶相および収量を示す。
Table 1. pH and concentration of {Mo72V30} in the precursor solution, and the product amount and their crystal phases after hydrothermal synthesis with or without the seed at 175°C for 20 ha.| Entry | pHb | Concentration of {Mo72V30}c | {Mo72V30} yieldd | Seede | Crystal structure of
the obtained solid | Amount of solid |
| 1 | 4.0 | 0.9 mM | 51% | —f | —g | 0.0 g |
| 2 | 3.4 | 1.0 mM | 57% | —f | Orth-MoVO | 0.3 g |
| 3 | 3.2 | 1.1 mM | 63% | —f | Orth-MoVO | 1.4 g |
| 4 | 2.7 | 1.0 mM | 57% | —f | Orth-MoVO | — |
| 5 | 2.2 | 0.6 mM | 35% | —f | Tri-MoVO | 5.0 g |
| 6 | 1.7 | 0.4 mM | 23% | —f | Hex-Mo0.87V0.13O2.94h+Tri-MoVO | — |
| 7 | 1.2 | 0.0 mM | 0% | —f | Hex-Mo0.87V0.13O2.94h | 6.7 g |
| 8 | 4.0 | 0.9 mM | 51% | Orth-MoVO | Orth-MoVO | 1.3 g |
| 9 | 4.0 | 0.9 mM | 51% | Tri-MoVO | Tri-MoVO | 1.7 g |
| 10i | 4.0 | 0.9 mM | 51% | Orth-MoVO | Orth-MoVO | 0.7 g |
| 11j | 4.0 | 0.9 mM | 51% | Tri-MoVWO | Tri-MoVO | 1.8 g |
| 12k | 4.0 | 0.0 mM | 0% | Orth-MoVO | —g | 0.0 g |
| 13 | 1.2 | 0.0 mM | 0% | Orth-MoVO | Hex-Mo0.87V0.13O2.94h | 6.9 g |
| a Hydrothermal synthesis was carried out using the precursor solution prepared by (NH6)Mo7O24·4H2O (Mo: 50 mmol) and VOSO4·5H2O (V: 12.5 mmol). b pH of the reaction mixture was changed from 1.0 to 4.0 by adding 2 M H2SO4 or 10 wt% ammonia. c Estimated by UV-Vis. d Based on V. e Seed amount was 0.3 g. f No seed was added. g No solid was formed. h JCPDF: 00–048–0766. i Unground Orth-MoVO seed was added. j Tri-MoVWO was used as a seed. k V2O5 was used as a V source. |
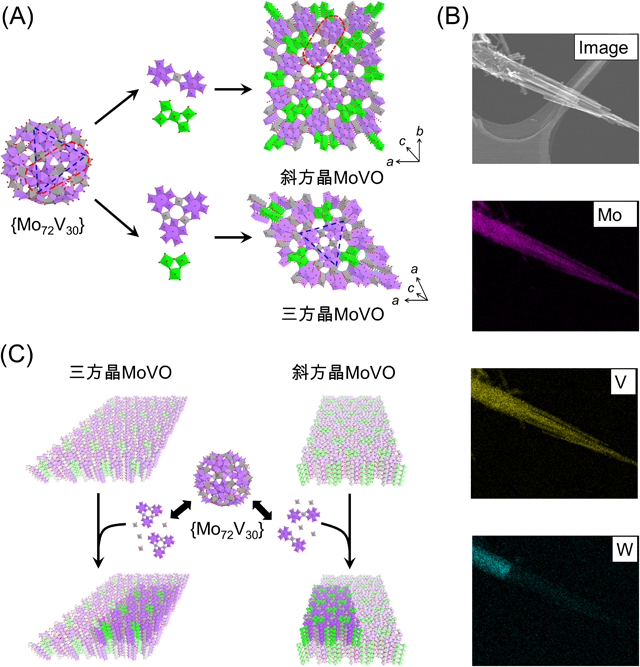
種結晶なしでは前駆体溶液のpHが3.4~4.0のときにはほとんど結晶は得られなかったが(Entry 1, 2), pHを3.2まで低下させると斜方晶MoVO(Orth-MoVO)が形成した(Entry 3)。pHを2.2までさらに低下させると,斜方晶MoVOと同様な組成,構造ユニットで構成されるがユニットの配列が異なる三方晶MoVO(Tri-MoVO)が形成した(Entry 5)。pHをさらに下げ,1.7とすると,金属酸素八面体のみから成り,構造内に{Mo6O21}6− 5員環ユニットを含まない擬六方晶系のMo0.87V0.13O2.94酸化物(Hex-Mo0.87V0.13O2.94)が三方晶MoVOとともに副生した(Entry 6)。pHを1.2とすると擬六方晶Mo0.87V0.13O2.94のみが得られた(Entry 7)。溶液UVにより前駆体溶液中の{Mo72V30}量を測定したところ,pH=2.7~4.0における{Mo72V30}量は0.9~1.1 mMほどであった。これは加えたVの51~63%が{Mo72V30}形成に関与していることを示している。pHを2.2まで下げると{Mo72V30}量は0.6 mMまで減少し,pHを1.2までさらに下げると同量はほとんど0となった。このとき,溶液中のポリ酸種としては{Mo72V30}ではなく,より縮合が進行した {Mo36O112}8−({Mo36})ユニットが形成していた。ここまでの結果を総合すると,{Mo72V30}存在下では{Mo6O21}6− 5員環ユニットを構造内に含む斜方晶および三方晶MoVOが形成し,{Mo72V30}非存在下では擬六方晶Mo0.87V0.13O2.94のみが得られた。{Mo72V30}と斜方晶MoVO,および三方晶MoVOはそれぞれ共通の構造パーツを有している。斜方晶MoVOでは2つの{Mo6O21}6− 5員環ユニットが一つの{VO6}リンカーで架橋した直線型のユニットであり,三方晶MoVOでは3つの{Mo6O21}6− 5員環ユニットが3つの{VO6}リンカーで架橋した三角形型のユニットである。この事実を鑑み,{Mo72V30}から供給されるビルディングユニットがそれぞれ縮合し結晶形成が進行するというユニット式結晶形成プロセスを提案した(Fig. 2(a))。高pH領域で{Mo72V30}が存在していてもMoVOが形成しなかった事実については,以降に概説する。
続いて,MoVO前駆体溶液中に粉砕した斜方晶MoVOや三方晶MoVOを種結晶として加えて水熱合成を行った20)。すると,本来MoVOが形成しないpH=4.0においても,種結晶と同様の結晶構造のMoVOが形成した(Entry 8, 9)。種結晶を粉砕せず,長い棒状結晶のまま用いると結晶収率は大きく低下したことから,棒状結晶を粉砕することで露出する結晶断面(a–b面)が結晶形成に強く関与していることが示唆された(Entry 10)。Wを含んだ三方晶MoVWOを種結晶として用いた場合,三方晶MoVOを種結晶とした場合とほぼ同収率で三方晶MoVOが得られた(Entry 11)。Fig. 2(b)に三方晶MoVWOを種結晶として合成したMoVOのSTEM-mapping像を示す。Mo,Vは棒状結晶に均一に分布していたが,Wは棒状結晶の真ん中までしか含まれていなかった。この時に合成したMoVOでは種結晶のみがWを含んでいることから,MoVOはMoVWO棒状結晶の断面部から形成していることが分かった。以上より,MoVOを種結晶として用いると,MoVO結晶断面部(a–b面)から新たなMoVOが形成すると結論した。一方,種結晶を用いた合成においても,{Mo72V30}は必須であった。同pH(pH=4),同Mo,V量で,V源にNH4VO3を用いて水熱合成を行ったところ(この時{Mo72V30}は形成していない),MoVOの結晶形成は観測されなかった(Entry 12)。また,{Mo72V30}の存在しないpH=1.2で水熱合成を行った場合,種結晶の添加の有無にかかわらず,{Mo6O21}6− 5員環ユニットを含まない擬六方晶系のMo0.87V0.13O2.94酸化物しか得られなかった(Entry 13)。以上の結果を総合すると,種結晶を用いたMoVO合成においては,種結晶の結晶断面(a–b面)が結晶核として機能し,{Mo72V30}が構造ユニット供給を担っていることが理解できる(Fig. 2(c))。pH=4.0で種結晶を用いない場合にMoVOが形成しなかった結果は,このpHにおいて{Mo72V30}が結晶核を形成できなかったことに起因すると考えられる。ゼオライト合成における種結晶添加法では,ゼオライト合成の前駆体溶液中に種結晶が有する構造ユニットが含まれているときに,新たなゼオライトが形成する21–23)。MoVOと{Mo72V30}が共通の構造ユニットを有していることを考えると,種結晶を用いたMoVO結晶形成プロセスはゼオライトのそれと類似している。金属酸素八面体をベースとしたMoVOとTO4四面体(T=Si,Alなどの元素)をベースとしたゼオライトが類似した機構で結晶形成する事実は大変興味深い。
以上,MoVOは前駆体溶液中に存在する{Mo72V30}から供給されるビルディングユニットを基盤としたユニット式の結晶形成プロセスにより形成することが明らかとなった。MoVO種結晶が存在すると,{Mo72V30}と共通の構造ユニットを有する種結晶断面部にビルディングユニットが集積し,新たなMoVOが形成していくことが分かった。
我々はこれまでに,水熱合成条件の制御,適切な熱処理により,同様の組成で構造ユニットの配列のみが異なる,斜方晶,三方晶,正方晶,および積層方向の配列は規則的であるが面方向が不規則な配列をしているアモルファスMoVOの合成に成功している(Fig. 1)。これらはいずれも,Mo,V金属を中心とした酸素八面体の複雑な配列により形成したa–b面が,c軸方向に頂点共有して積み重なった層状構造体である。a–b面は,{Mo6O21}6− 5員環ユニットが酸素八面体を介してネットワーク状に連結し,斜方晶,三方晶,およびアモルファスでは6,7員環が形成され,正方晶では5,6員環が形成する。Fig. 1に示したこれらの結晶構造の妥当性は,HAADF-STEM像から明らかである。Table 2にこれら4つの結晶のN2吸着測定および触媒反応結果を示す。4つの結晶について,100 nm2当たりの7員環数を結晶構造モデルやHAADF-STEM像から見積もったところ,斜方晶MoVOでは73個,三方晶MoVOでは68個,アモルファスMoVOでは10~30個,正方晶では0個であった。これらを用いて窒素吸着測定を行ったところ,7員環を有する斜方晶,三方晶,アモルファスはミクロ細孔吸着を示したが,正方晶では示さなかった。このことから構造中の7員環がミクロ細孔として機能していることは明らかである。
Table 2. Relationship between micropore and catalytic activity.| Catalyst | Elemental
compositiona(V/Mo) | Number of 7-membered ring/100 nm2 | External surface areab/m2 g−1 | Micropore volumeb/cm3 g−1 | Ethane
conv.c/% | ACR
conv.d/% |
| Orth-MoVO | 0.38 | 73 | 7.2 | 14.0 | 42.2 | 53.8 |
| Tri-MoVO | 0.32 | 68 | 18.0 | 4.0 | 25.2 | 99.8 |
| Tet-MoVO | 0.38 | 0 | 2.7 | 0 | <1 | <1 |
| Amor-MoVO | 0.38 | 10~30 | 5.7 | 2.8 | 5.9 | 9.7 |
| a Determined by ICP. b Measured by N2 adsorption at liquid N2 temperature and estimated by t-plot method. c Reaction condition: catalyst amount, 0.5 g; reaction gas feed, C2H6/O2/N2=5/5/40 ml min−1; reaction temperature, 313~319°C. d Reaction condition: catalyst amount, 0.25 g, reaction gas feed, ACR/O2/H2O/N2/He=2.5/8.0/27.1/39.5/30.5 ml min−1; reaction temperature, 217~218°C. ACR represents acrolein. |
これらを用いてエタンおよびアクロレイン選択酸化反応を行った。斜方晶,三方晶,アモルファスはエタンおよびアクロレインの転化に活性を示したが,正方晶では活性を示さなかった(Table 2)。エタン,アクロレイン選択酸化反応において触媒活性の強い構造依存性が見て取れた。これらの触媒の違いは構造中の7員環の有無のみであったことから,7員環がエタン,アクロレインの転化に関与していることは明らかであろう。
エタン選択酸化活性は斜方晶MoVO>三方晶MoVO>アモルファスMoVO≫正方晶MoVOであった。この序列は触媒の表面積(斜方晶:7.2 m2 g−1,三方晶:18.0 m2 g−1,正方晶:2.7 m2 g−1,アモルファス:5.7 m2 g−1)や100 nm2当たりの7員環数(斜方晶:73個,三方晶:68個,正方晶:0個,アモルファス:10~30個)と関係せず,N2吸着測定から求めた7員環に由来するミクロ孔容積(斜方晶:14.0 cm3 g−1,三方晶:4.0 cm3 g−1,正方晶:0 cm3 g−1,アモルファス:2.8 cm3 g−1)の序列と一致していた。これはエタンが7員環表面でなく7員環細孔内で転化していることを示唆している。一方でアクロレイン選択酸化活性は三方晶MoVO>斜方晶MoVO>アモルファスMoVO≫正方晶MoVOであり,エタン酸化活性とは異なる序列となった。この活性の序列は触媒の表面積と100 nm2当たりの7員環の数を考えるとよく理解できる。この事実はアクロレインが7員環の表面で転化していることを示唆している。これらの詳細については次項で詳しく解説する。
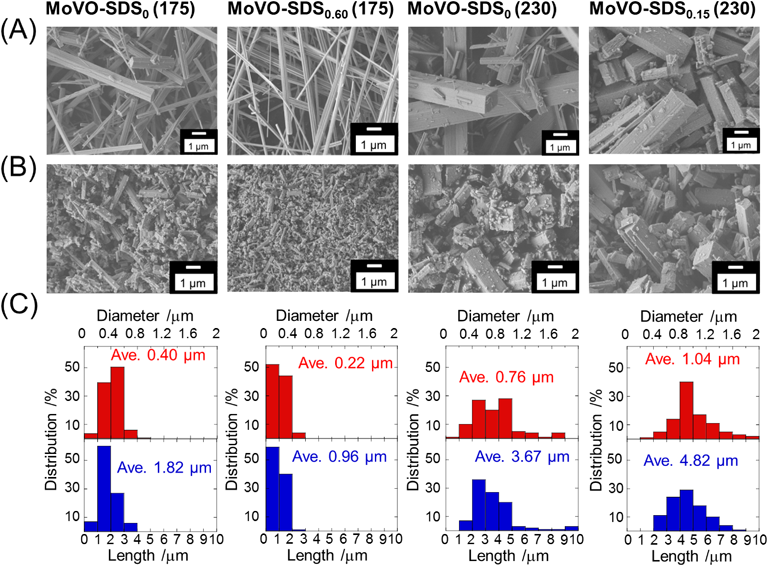
4つの異なる結晶相のMoVOの触媒活性を比較することで,エタンとアクロレインはMoVOの7員環部位で転化することは明らかである。一方,エタンとアクロレインでは基質活性化部位が異なることが示唆されている。エタン,アクロレイン選択酸化反応における基質活性化部位を明らかにするため,ミクロ細孔容積が同じで外表面積のみの異なる種々のMoVOを合成し,触媒活性の外表面積依存性を検討した。もし基質が7員環表面で転化してれば,触媒活性は外表面積の変化に依存するはずである。一方,基質がミクロ細孔内で転化しているとすれば,活性は外表面積に依存しないはずである。MoVOの外表面積を制御するため,界面活性剤(Sodium dodecyl sulfonate, SDS)の添加や水熱合成温度の制御を行うことで,結晶核形成速度および結晶成長速度を変化させ,MoVOの結晶サイズ制御を試みた。SDSの添加量は仕込み比でSDS [mol]/(Mo+V)[mol]=x (x=0, 0.15, 0.30, 0.60)とし,合成温度は175°Cまたは230°Cとした。以下,合成した触媒をMoVO-SDSx (175 or 230)で表す。合成した試料はいずれも同様の組成のMoVOであった。これらの触媒の諸物性(組成,結晶構造,外表面積等)は反応前後でほとんど変化しなかったため,以下のキャラクタリゼーションは触媒反応の影響を受けた後の,反応後の試料について行った。Fig. 3にエタン反応後の代表的な試料のSEM像および各棒状結晶の軸径,軸長の平均値を示す。
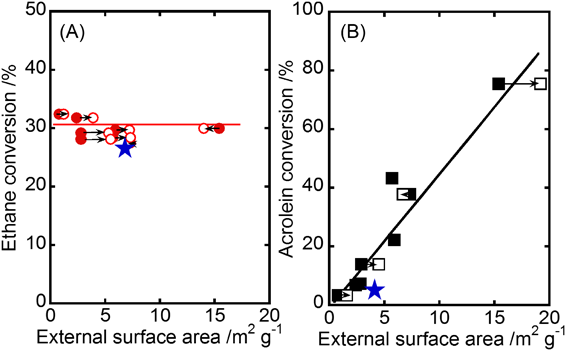
Fig. 3に示すように,SDSの添加や水熱合成温度の制御によって,MoVOの結晶サイズを大きく変化させることができた。175°Cで合成した試料では,SDSを過剰に転化したとき(MoVO-SDS0.60 (175)),棒状結晶の軸径平均(100個の結晶を測定)はSDSを添加していない場合の0.40 µmから0.21 µmまで減少した。230°Cで合成した試料は,軸径平均が0.76 µmへと大きく増大した(MoVO-SDS0 (230))。この温度でx=0.15となるようにSDSを添加すると(MoVO-SDS0.15 (230)),軸径平均は1.04 µmへとさらに大きく増大した。一方,これらの結晶の軸長に対する軸径の比(アスペクト比)は結晶サイズに関係なく,ほとんど同様であった。この事実は,合成した試料が相似形であることを示している。以上,SDSの添加や合成温度の制御により,結晶核生成速度や結晶成長速度が変化し,相似形で異なるサイズの結晶が得られたと結論した。結晶サイズの変化による外表面積の変化はN2吸着測定の結果からも明らかであり,1.2~14.0 m2 g−1の範囲で触媒の外表面積を制御することができた。続いて,エタン酸化反応後の各MoVOに対しエタン吸着測定を行ったところ,結晶サイズに関係なく,これらはいずれもほぼ同様のエタン吸着量を示した(16.1~18.8×10−3 cm3 g−1)。結晶構造から見積もられるミクロ細孔容積の理論値は22.4×10−3 cm3 g−1である。観測されたエタン吸着量は理論値と比べて若干小さい値となったが,それでも合成した試料がエタンを吸着できる7員環を有することは明らかである。以上の実験から,合成した試料は結晶構造,組成,結晶形状,およびミクロ細孔容積がほぼ同様で外表面積のみが異なるMoVOであると結論した。Fig. 4にこれらが示したエタンおよびアクロレイン転化率と触媒の外表面積との関係を示す。触媒反応条件はFig. 4の脚注に示した。生成物選択性は各MoVOで変化はなく,エタン選択酸化反応では,エチレンが90%程度の選択率で生成し,残りは酢酸,COxであった。アクロレイン選択酸化反応では95%程度の選択率でアクリル酸が生成し,残りは酢酸,COxであった。
アクロレインを基質に用いた場合,アクロレイン転化率は触媒の外表面積に大きく依存した。この事実は,アクロレインが触媒表面で転化していることを示している。MoVO-SDS0 (175)に粉砕処理を行わなかった場合(MoVO-SDS0 (175, ng)),アクロレイン転化率は大きく減少した。この事実は粉砕処理によって露出した結晶断面(a–b面)がアクロレインの転化に寄与していることを示している。前述した4つの結晶相におけるアクロレイン選択酸化活性の比較も考慮し,アクロレインは触媒表面に位置する7員環で転化していると結論した。反応性の高いアルデヒド基が7員環表面に効率的にトラップされ,活性化されたと考えられる。一方,エタンを基質に用いた場合,エタンの転化率は触媒の外表面積と関係なく,ほとんど同様であった。これはエタンが触媒表面で転化していないことを意味している。前述した4つの結晶相におけるエタン選択酸化活性の比較から,エタンの転化に7員環が必須であることは明らかである。一方,MoVO-SDS0 (175, ng)がMoVO-SDS0 (175)とほぼ同様の活性を示したことから,粉砕処理により露出したa–b面はほぼ反応に関与しないことが分かる。さらに,本実験で用いた反応条件において,7員環が異元素で占有されている触媒がほとんど活性を示さないことも別途実験により明らかにしている24)。また,拡散の影響を評価するために,結晶サイズの大きく異なる3つの触媒(軸径平均:0.21 µm,0.40 µm,1.04 µm)を用いて触媒活性の接触時間依存性を評価した。その結果,これらの触媒の活性はいずれの接触時間においてもほとんど同様であった。以上の結果から,MoVOは7員環ミクロ細孔内で拡散の影響なくエタンを活性化し,エチレンを与えることが明らかになった。ここまでで得られた結果は,エタン選択酸化反応において,7員環ミクロ細孔内が7員環表面に比べて明らかに優位な触媒反応場として機能していることを示している。これにはミクロ細孔中の電場の勾配による基質のC–H結合分極や,基質濃度の部分的な増大が関与していると思われる25–26)。
5. 7員環周りの局所構造とエタン選択酸化活性の関係15–16)
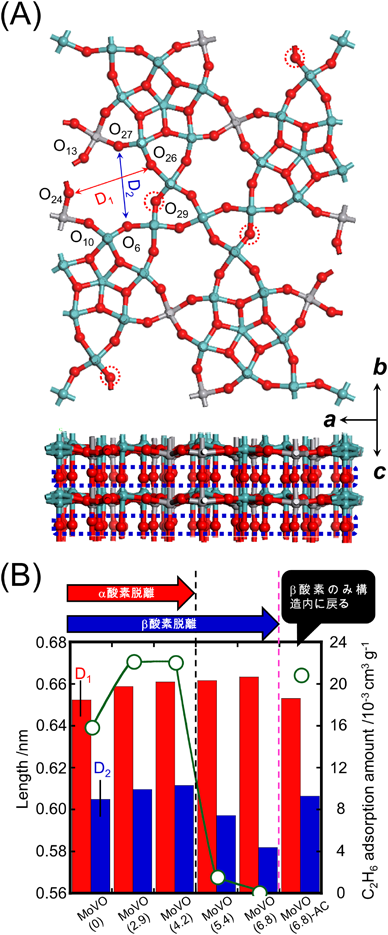
これまでの研究から,MoVOの7員環ミクロ細孔径が酸化還元処理により,連続的に,かつ可逆的に制御可能であることが明らかになっている。ここまでで7員環はエタンを内部に取り込み活性化していることは明らかなので,7員環細孔径はエタン酸化活性に大きく影響することが考えられる。そこで,MoVOに酸化還元処理を行うことによってミクロ細孔性質を変化させ,ミクロ細孔性質がエタン選択酸化活性に及ぼす影響を検討した。この目的のため,構造ユニット内からの格子酸素脱離量が異なる種々MoVOを調製し,キャラクタリゼーションを行った。格子酸素脱離量の測定にはTPR, TGを用いた。以下,調製した試料をMoVO (δ)(δはMo29V11O112からの酸素脱離量)とする。MoVO (0)は水熱合成後のMoVOを400°Cで2 h空気焼成を行うことで得た。還元処理後に同じ条件で空気焼成を行うことで得た試料は,試料名の末尾にACを付けて表記している。酸化還元処理による不純物の生成は,XRD,IR,およびRaman測定では観測されなかった。種々解析の結果,還元によって優先的に構造から脱離し,再酸化しても構造内に戻らない格子酸素と,還元によって連続的に脱離し,可逆的に構造内に戻る格子酸素が存在することが分かった。以下,前者をα酸素,後者をβ酸素とする。TPRスペクトルから,MoVO (4.2)までの還元でα酸素は構造から脱離し終えることが示されている。Fig. 5にMoVOの構造モデル,Rietveld解析によって得られた構造モデルから算出した7員環ミクロ細孔の長軸径,短軸径,および種々還元度の異なるMoVOのエタン吸着量を示す。種々解析の結果,α酸素は7員環に面したペンタマーユニット内の架橋酸素,β酸素はc軸に結合したアキシャル酸素であることが分かった。エタン吸着量はMoVO (0)~MoVO (4.2)では15.8~22.1×10−3 cm3 g−1であり,有意な差は観測されなかった。一方,MoVO (5.4)以上に還元した試料では,エタン吸着がほとんど観測されなかった。Rietveld解析後の構造モデルから算出した7員環ミクロ細孔径について,7員環長軸径は還元に伴って連続的に増大した。一方,7員環短軸径はMoVO (4.2)までの還元では連続的に増大したにもかかわらず,以降の還元で大きく減少した。
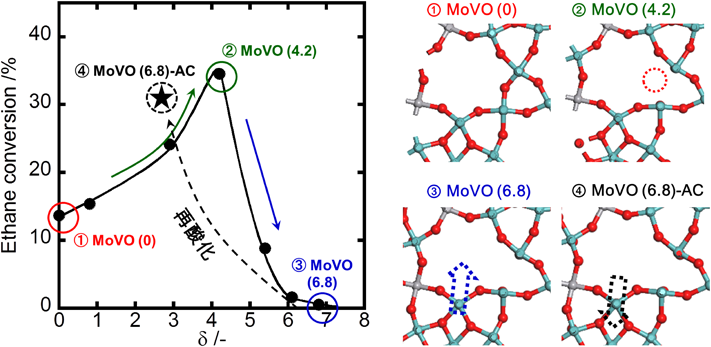
XRD,IR,Raman測定により,MoVO (5.4)以上の還元で,{Mo6O21}6− 5員環ユニットの膨張に伴うa–b面の局所構造変化が観測された。このことから,MoVO (5.4)以上の還元によって観測されるエタン吸着量の減少は,{Mo6O21}6− 5員環ユニットが膨張することで7員環が圧迫され,エタンが7員環細孔内にアクセスできなくなったことによると結論した。再酸化処理を行ったMoVO (6.8)-ACでは,7員環細孔径は還元前のMoVO (0)とほぼ同様となった。実際,IR,Raman測定でも,再酸化処理によって{Mo6O21}6− 5員環ユニットが収縮することが示されている。再酸化処理を行うとα酸素は構造内に戻らないまま,{Mo6O21}6− 5員環ユニットが収縮することで7員環径が増大すると結論した。以上の実験により,還元度の異なる種々MoVOの構造モデルが得られた。Fig. 6に還元度の異なるMoVOを用いた反応開始10 min時点でのエタン転化率とδの関係を示す。
MoVO (0)のエタン転化率は13%程度であった。還元処理を行ったMoVO (2.9)では,エタン転化率は24%程度に増大した。さらに還元が進行したMoVO (4.2)ではエタン転化率は35%程度へと大きく増大した。しかし,これ以上の還元により,MoVO (5.4)では9%程度,MoVO (6.1),MoVO (6.8)ではほとんど0%と,エタン転化率は大きく減少した。エタン吸着実験から,MoVO (5.4) 以上の還元によって7員環細孔径が収縮し,エタンが細孔内にアクセスできなくなることが分かっている。エタンは7員環細孔内で転化することから,MoVO (5.4)以上の還元で観測されたエタン酸化活性の減少は,エタンが細孔内にアクセスできなくなったことに起因する。MoVO (6.8)-ACのエタン転化率はMoVO (0)よりもはるかに高く,31%程度であった。MoVO (0) とMoVO (6.8)-ACでは結晶構造,組成,および酸化状態がほぼ同じであり,両者の違いはα酸素の有無のみである。このことから,α酸素が脱離することでエタン酸化活性が大きく増大することが明らかであろう。MoVO (0) とMoVO (6.8)-ACを用いてアレニウスプロットを取ったところ,α酸素の脱離は活性化エネルギーに影響せず,反応頻度因子のみを増大させることが分かった。この結果はα酸素が脱離した構造がエタン転化における活性点構造であることを示している。以上の結果を総合し,MoVOを用いたエタンの酸化に関してFig. 7に示す反応スキームを提案した。
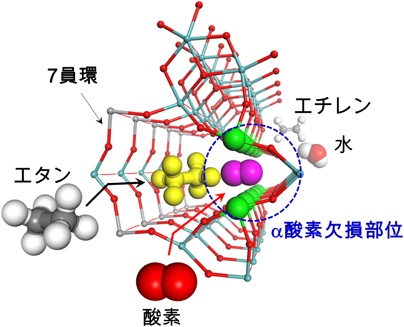
エタンと酸素は反応中,同時にミクロ細孔に取り込まれる。この時,α酸素欠損部位が存在するとエタンからのC–H引き抜きに活性な酸素種が形成し,効率的にエタンがエチレンへ転化する。以上より,7員環近傍の局所構造変化とエタン選択酸化反応における触媒活性の関係が明らかになった。
結晶性Mo3VOx複合酸化物(MoVO)の触媒調製から触媒反応に至るまで,最近の進歩を概説した。MoVOは水熱条件下,巨大ボール型ポリオキソメタレートから供給されるビルディングユニットの自己組織化によって洗練された結晶構造を形成する。その結晶構造中には低級アルカンがちょうど取り込めるほどの大きさのミクロ細孔が形成し,5つの金属酸素八面体で構成されるペンタマーユニットがこれに隣接する。エタン選択酸化反応においては,エタンはミクロ細孔中に取り込まれ,細孔中の電場勾配によりC–H結合が分極する。同時に,ミクロ細孔に隣接したペンタマーユニット内に形成している酸素欠陥が分子酸素を適度に活性化し,活性化されたエタンから水素原子を引き抜く。このように,MoVOの極めて高い触媒機能はミクロ細孔に隣接した触媒活性点が高密度に周期的に配列していることに由来し,これは見事なまでに緻密に制御された結晶構造によってはじめて成し遂げられる。ナノスケールな視点で触媒反応を俯瞰すると,高活性な触媒創出のためには触媒活性点の構造設計が決定的に重要と思われる。触媒設計論は議論百出であろうが,結晶構造をベースとした議論は常に心に留めておくべきであろう。
引用文献References
1) T. Katou, D. Vitry, W. Ueda, Chem. Lett., 32, 1028 (2003).
2) W. D. Pyrz, D. A. Blom, M. Sadakane, K, Kodato, W. Ueda, T. Vogt, D. J. Buttrey, Chem. Mater., 22, 2033 (2010).
3) T. Lunkenbein, F. Girgsdies, A. Wernbacher, J. Noack, G. Auffermann, A. Yasuhara, A. K. Hoffmann, W. Ueda, M. Eichelbaum, A. Trunschke, R. Schlögl, M. G. Willinger, Angew. Chem. Int. Ed., 54, 6828 (2015).
4) M. Sadakane, K. Kodato, T. Kuranishi, Y. Nodasaka, K. Sugawara, N. Sakaguchi, T. Nagai, Y. Matsui, W. Ueda, Angew. Chem. Int. Ed., 47, 2493 (2008).
5) M. Sadakane, S. Ohmura, K. Kodato, T. Fujisawa, K. Kato, K. Shimidzu, T. Murayama, W. Ueda, Chem. Commun., 47, 10821 (2011).
6) W. Ueda, J. Jpn. Petrol. Inst., 56, 122 (2013).
7) F. Wang, W. Ueda, Appl. Catal., A, 346, 155 (2008).
8) N. Watanabe, W. Ueda, Ind. Eng. Chem. Res., 45, 607 (2006).
9) C. Qiu, C. Chen, S. Ishikawa, T. Murayama, W. Ueda, Top. Catal., 57, 1163 (2014).
10) C. Chen, K. Nakatani, T. Murayama, W. Ueda, ChemCatChem., 5, 2869 (2013).
11) T. Konya, T. Katou, T. Murayama, S. Ishikawa, M. Sadakane, D. Buttrey, W. Ueda, Catal. Sci. Technol., 3, 380 (2013).
12) M. Sadakane, N. Watanabe, T. Katou, Y. Nodasaka, W. Ueda, Angew. Chem., Int. Ed., 46, 1493 (2007).
13) S. Ishikawa, X. Yi, T. Murayama, W. Ueda, Appl. Catal., A, 474, 10 (2014).
14) S. Ishikawa, X. Yi, T. Murayama, W. Ueda, Catal. Today, 238, 35 (2014).
15) S. Ishikawa, D. Kobayashi, T. Konya, S. Ohmura, T. Murayama, N. Yasuda, M. Sadakane, W. Ueda, J. Phys. Chem. C, 119, 7195 (2015).
16) S. Ishikawa, W. Ueda, Catal. Sci. Technol., 6, 617 (2016).
17) M. Sadakane K. Endo, K. Kodato, S. Ishikawa, T, Murayama, W. Ueda, Eur. J. Inorg. Chem., 10–11, 1731 (2013).
18) R. Canioni, M. Roch, L. Laronze, M. Haouas, F. Taulèlle, J. Marrot, S. Paul, C. Lamonier, F. Paul, S. Loridant, M. M. J. Millet, E. Cadot, Chem. Commun., 47, 6413 (2011).
19) M. S. Sanchez, F. Girgsdies, M. Jastak, P. Kube, R. Schlögl, A. Trunschke, Angew. Chem., Int. Ed., 51, 7196 (2012).
20) S. Ishikawa, M. Tashiro, T. Murayama, W. Ueda, Cryst. Growth Des., 14, 4553 (2014).
21) K. Itabashi, Y. Kamimura, K. Iyoki, A. Shimojima, T. Okubo, J. Am. Chem. Soc., 134, 11542 (2012).
22) K. Iyoki, K. Itabashi, W. Chaikittisilp, S. P. Elangovan, T. Wakihara, S. Kohara, T. Okubo, Chem. Mater., 26, 1957 (2014).
23) S. Goel, S. I. Zones, E. Iglesia, Chem. Mater., 27, 2056 (2015).
24) S. Ishikawa, T. Murayama, M. Kumaki, M. Tashiro, Z. Zhang, A. Yoshida, W. Ueda, Submitted.
25) G. Sastre, A. Corma, J. Mol. Catal. A -Chem., 305, 3 (2009).
25) C. M. Z. Wilson, A. Corma, P. Viruela, J. Phys. Chem., 98, 10863 (1994).