光エネルギーの有効活用は,エネルギー問題解決に向けた高効率な光触媒,太陽電池,人工光合成への応用の観点から,注目が集まっている。光エネルギーを化学エネルギーへと変換する光触媒・光合成システムには,光によって生成した励起子を効率よく外部に取り出す構造を創製する必要がある。無機と有機をナノレベルで精密に配置したナノ構造体は,ホスト–ゲスト相互作用による機能発現が期待され,吸着材,触媒,化学・生体センサー,光学材料や発光材料などへの応用に向けて多くの興味を集めてきた。無機酸化物をホスト材料として用いる場合には,無機酸化物に特有の高い熱的・機械的安定性をゲスト分子に付与するだけではなく,無機酸化物が形成する精密かつ特異な空間を活用することで新たな機能を創り出すことができる。特に,光エネルギーにより生成した励起子の緩和過程を操作する手法として,固体が創り出す表面や空間の利用は,実用的な観点からも重要である。近年のメソポーラスシリカを契機とした有機化合物を利用した無機化合物のナノ構造制御技術は,無機酸化物の表面や空間を精密に設計し,分子の光物理的および光化学的性質をつかさどることができるようになりつつあり,研究領域を広げている。
ホスト材料としてゼオライトなどのシリカ材料は,熱的・化学的に安定であり,240 nm以上の光に対して高い透過性を有し,かつナノ細孔構造が,分子やイオンを規則的に導入することが可能なことから,光機能を精緻に制御したナノハイブリッド系を構築することが期待できる。シリカナノ構造中への発光分子・イオンの導入は,レーザー発光モジュールやディスプレイ材料などの光機能材料への応用に向けて,これまでにも多くの研究が行われてきた。これらは,Turro1)やRamamurthy2)らの研究から始まり,規則性メソポーラス有機シリカ3)などへ展開されてきている。特に,ゼオライト細孔内に有機分子とともに組み込むことによりNd(III)からの近赤外フォトルミネッセンスを可能とする4),希土類と光増感分子をゼオライト細孔内に組み込むことによりRGB同時発光を可能とする5)などの精密なナノ空間を利用した研究が報告されている。これらは,紙幅の都合により詳細を述べることはできないが,これまでにも多くのレビューや解説記事があるので参照していただきたい。
また,層状ケイ酸塩のような層状無機酸化物は,二次元に広がった空間を提供し,ゲスト種が二次元に束縛された状態にあり,ゼオライトなどの三次元細孔とは異なる特異な環境に由来した光エネルギー緩和過程の制御が行われてきた6)。特に,層状化合物上のゲスト分子の凝集現象・分離現象を制御し,光化学物性を制御する試みが行われてきている。小川らは,ルテニウム錯体–粘土鉱物複合体の発光特性およびルテニウム錯体の凝集制御について報告している7)。ポリビニルピロリドン(PVP)を予め導入した合成フッ素四ケイ素雲母(TSM)をホストに用いたところ,PVPとRu(bpy)32+が層間に共存した層間化合物が合成され,この特異な層間環境によりルテニウム錯体の発光ピークが溶液中の発光ピークと比べ低波長に観測された。また,ポルフィリン分子を層間に導入した粘土–ポルフィリン複合体では,ポルフィリンを高担持量で粘土表面上に吸着させても発光効率の低下を招く自己消光現象は観測されないという特異な性質を示した8)。このサポナイト–ポルフィリン複合体が人工の光捕集システムに適するようポルフィリンの選択,吸着密度,ドナー–アクセプター比の最適化を行ったところ,100%に近いエネルギー移動効率を達成したことを報告した。これらのように,層状化合物へのゲスト分子の凝集・分離現象を精密に制御することで,新たな光機能を付与できると見込まれる。
以上の事例は,無機ナノ材料が創り出す空間を精密に利用することで特異な光励起緩和過程を制御することに成功した例である。本稿では,近年筆者らが報告してきたナノ空間を利用した光励起緩和過程の制御についてまとめた。前半部分は,ゼオライトの細孔内や層状ケイ酸塩層間にイリジウム錯体を導入し,シリケート骨格が錯体の光励起状態の緩和過程,すなわち発光および電子移動に与える影響を明確にする。発光に関しては,同じゼオライト粒子内で二種類のイリジウム錯体を配列させ,同時に発光させることを試みる。光誘起電子移動反応において,ゼオライト粒子内で錯体と電子受容体を配列させ,溶液中の電子受容体への電子移動反応の促進を狙う。後半部分では,新たにチオール–エンクリック反応(2.2参照)を利用したチタン酸ナノシートとタングステン酸ナノシートの交互積層体を合成し,ナノシート間で起こる光誘起電子移動を扱う。本手法で得られる交互積層体は,層間に導入するアルキル鎖炭素数を変化させることで,ナノシート間の距離のナノメートルオーダーでの精緻な制御が可能である。ナノシート間の距離が,チタン酸ナノシート伝導帯からタングステン酸ナノシート伝導帯への紫外光誘起の電子移動反応に及ぼす影響を明確にする。更に,交互積層体の層間に色素分子を導入することで,色素分子を介したタングステン酸ナノシートからチタン酸ナノシートへの可視光誘起の電子移動反応が起こることも示す。
2. シリカナノ空間に閉じ込めたイリジウム錯体の励起状態制御
2.1 3次元マイクロポーラス内に閉じ込めたイリジウム錯体
FAU型ゼオライトの細孔内では,ルテニウム錯体などのトリスビピリジン金属錯体の合成が行われてきた1),2)。これは,トリスビピリジンルテニウム錯体は,その分子サイズが直径およそ1 nmであり,FAU型ゼオライトのスーパーケージの直径(約1.3 nm)とほぼ同程度あるため,スーパーケージ一つに対し,錯体分子を一個だけ入れることができるためである。また,ルテニウム錯体を溶液からイオン交換で導入することは,FAU型ゼオライトのスーパーケージの入り口のサイズが0.7 nmなので不可能であるため,錯体の中心金属と配位子分子をゼオライトの細孔内に導入し,細孔内で配位結合を形成させるship-in-a-bottle合成法が開発されてきた。我々は,ship-in-a-bottle合成法を利用し,トリスオルトメタル化イリジウム錯体をゼオライト細孔内で合成し,光誘起励起状態の制御を試みた9),10)。
イリジウム錯体は,イリジウムの重原子効果により,一重項からの項間交差を促進し,励起寿命の長い三重項励起状態を生成する。この三重項励起状態は,量子効率高く発光するため,有機EL材料などの発光材料としての利用が期待されている。イリジウム錯体の三重項励起状態は,配位子のπ共役系が広がりを変化させることで制御でき,配位子の構造を変えることにより,発光色や励起エネルギー準位を調整できる。また,イリジウム錯体を水の還元による水素生成光触媒として用いることで,従来のルテニウム錯体より量子収率が高いことが報告されている。イリジウム錯体は,励起状態で強い還元力を持つことから,有機化合物の光還元反応に応用されている。このような光還元反応を効果的に進行させるためには,励起状態の失活過程を抑制する必要がある。励起状態の錯体から電子受容体へ電子移動させ,電荷分離させることは,失活過程を抑制する有効な手法の一つであり,錯体と電子受容体とを空間的に精密に配置することが重要な因子となる。我々は,ゼオライト細孔内に固定化されたイリジウム錯体からゼオライト粒子の外部に電荷分離させることが,光エネルギーを化学エネルギーへと変換させるための理想的な環境を提供すると考えた。
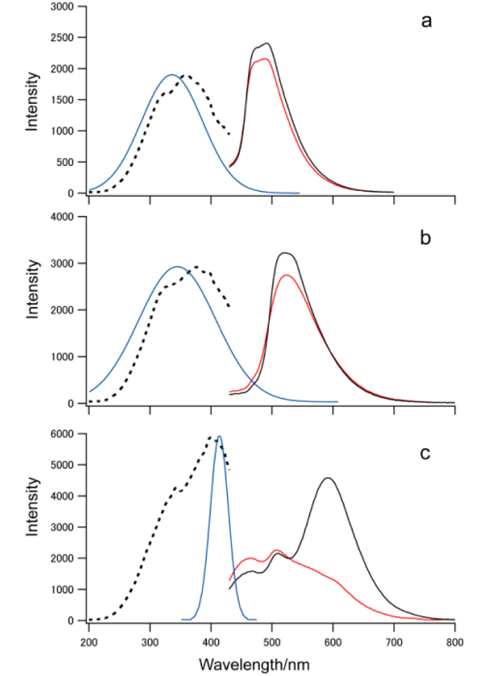
イリジウム錯体をゼオライト細孔内に導入するために,イリジウムイオンをあらかじめ細孔内にイオン交換させ,そこに配位子としてphenylpridine (ppy)および4′,6′-difluorophenylpyridine (dfppy)を導入した。その後,マイクロ波加熱を用い,X型ゼオライトのスーパーケージ内でイリジウム錯体をship-in-a-bottle合成し,Ir(ppy)3-XおよびIr(dfppy)3-Xを得た。Ir(ppy)3-X,Ir(dfppy)3-Xの紫外可視光拡散反射吸収スペクトルはそれぞれ330–400 nm付近に錯体のMLCTに帰属される吸収を示し,発光スペクトルにおいて,それぞれの生成物のピークトップは520,490 nmであった(図1(a)および(b))。この結果は,イリジウム錯体の生成を示している。一方で,よりπ電子系の広い2-phenylquinoline (phq)を配位子として反応させた場合,洗浄により拡散反射吸収および発光強度が減少することが確認された(図1(c))。この減少は,Ir(phq)3錯体がゼオライト粒子の外表面で生成したことを示している。これは,分子サイズの大きいphqがゼオライトの細孔内に導入できず,外表面で生成するためであり,Ir(ppy)3-X,Ir(dfppy)3-Xは,ゼオライトのスーパーケージ内に閉じ込められている状態であることが分かる。
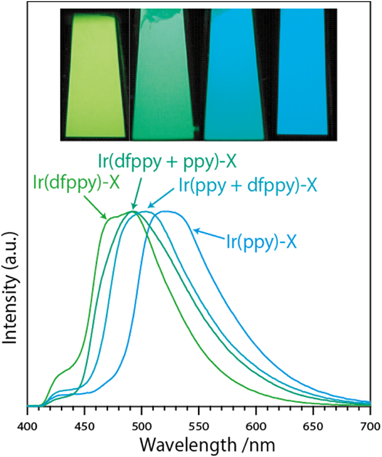
さらに,配位子dfppyとppyを順次反応させることにより,二種類のIr錯体Ir(dfppy)3とIr(ppy)3をゼオライト細孔内で共存させた。単独種のIr錯体導入ゼオライトと合わせ,これらのサンプルは青色から緑色へ種々の発光色を出すことができた(図2)。また,ppyとdfppyを同時にIr3+交換ゼオライトへ導入・錯体化させ,その際ppyとdfppyの比率を変えることにより,青色から緑色への発光色が変わるイリジウム錯体–ゼオライトの構築に成功した。この細孔内に導入したイリジウム錯体は,室温で比較的高い発光量子収率を示した。
イリジウム錯体の励起状態を化学変換系へ利用するため,ゼオライト細孔内のイリジウム錯体からビオローゲン類縁体の光還元を行い,光照射により得られた励起電子を溶液中に取り出すことを試みた10)。ビオローゲン類縁体は,ラジカルカチオンが安定に存在し,そのラジカルカチオンが可視光領域に吸収を持つため,容易に検出される。また,分子構造を設計することでその電荷を制御することができる。Ir(ppy)3-Xを双性イオンで中性のビオローゲン類縁体であるpropyl viologen sulfonate (PVS)と水溶液中に共存させた溶液に光照射することで光誘起電荷分離挙動を検討した。370 nmの励起光による520 nmの緑色発光が,PVSの添加量の増加に伴い消光し,溶液中にPVSの還元種であるラジカルアニオンが観測されたことから,イリジウム錯体からPVSへの電子移動が確認された。この電子移動による消光速度は,溶液での電子移動に比べておよそ300倍程度遅くなっていた。これは,イリジウム錯体がゼオライト細孔内に取り込まれることで,溶液中と比べてPVSと近接する頻度が減少したためと考えている。そこで,電子移動の効率を上げるため,Ir(ppy)3-Xに対して,カチオン性のビオローゲン類縁体であるジクワットイオン(2DQ2+)を反応させ,Ir(ppy)3が導入されていない細孔内へ2DQ2+を導入した。2DQ2+を導入することにより,PVSラジカルアニオンの生成速度は,導入前に比べて1.9倍速くなった。これは,2DQ2+がゼオライト粒子内部のIr(ppy)3錯体と外部のPVSの電子移動をリレーする役割を担いIr(ppy)3から溶液中の電子受容体への電子移動を促進させたことを意味している。光励起した電子を,非効率な逆電子移動反応を抑制し,電荷分離状態を形成することは,太陽エネルギーを蓄えるための分子集合体の構築において,ゼオライトナノハイブリッドが重要な役割を果たす可能性を示している。電荷の空間的分離は逆電子移動を抑えるための有効な手段であり,反応物や生成物を選別する能力を有するゼオライトと複合化することで,より理想的な環境が提供されると考える。
2.2 2次元空間内に閉じ込めたイリジウム錯体
層状ケイ酸塩はゲスト分子を配列させるのに適したホスト物質である。また,Ir(ppy)3のppy配位子の一つをビピリジン(bpy)配位子と置換したIr(ppy)2(bpy)+錯イオンは周囲環境に応じ,錯体の幾何構造が変化し,発光特性が変化する性質を持つ。その結果,層状ケイ酸塩が提供する二次元空間へ配列されたIr(ppy)2(bpy)+錯イオンは,層状ケイ酸層間の環境を反映した発光特性を示すことがわかった11)。
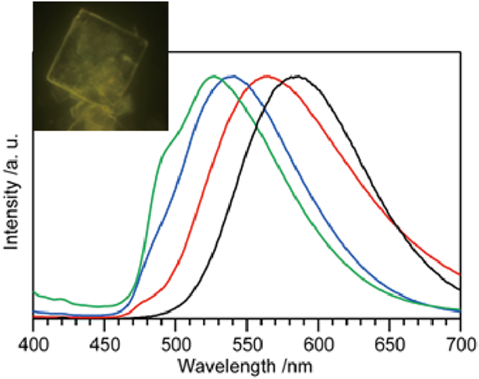
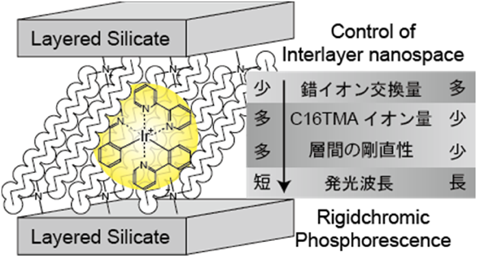
層状ケイ酸塩の層間に存在するナトリウムイオンをヘキサデシルトリメチルアンモニウム(C16TMA)イオンとイオン交換し,層間を拡張した層状ケイ酸塩のC16TMAイオン交換によりIr(ppy)2(bpy)+錯イオンを導入した。錯イオン導入量は,イオン交換の温度条件(室温,82°C,120°C)により制御可能であり,それぞれ300,60,20 nm3あたり錯イオン一個が導入された。また錯イオン周囲には大量のC16TMAイオンが残存する環境であった。そのため,錯イオンは孤立して存在し,濃度消光などの錯イオン同士の相互作用は無視できる状況にあると予想した。導入量の少ないイリジウム錯イオン–層状ケイ酸塩複合体の発光スペクトル(図3)は,溶液中の発光スペクトルと比較し,大きく短波長シフトすることが確認された。また,複合体の発光スペクトルは,イリジウム錯体導入量が増えるにつれて溶液中の発光スペクトルに近い長波長側へとシフトした。
発光スペクトルのシフトが起こるメカニズムとしては,層間の剛直性がイリジウム錯体の発光過程を変化させたと考察した(図4)。イリジウム錯イオンは,室温下溶液中でMetal to Ligand Charge Transfer(3MLCT)から発光するが,77 Kで凍結させたマトリックス中では,Ligand to Ligand Charge Transfer(3LLCT)から大きな低波長シフトを伴って発光する。層間拡張に用いたC16TMAイオンと錯イオンとのイオン交換量が少ない場合には,シリケートの層と層の間にC16TMAイオンが密に存在しているため,層間環境が剛直となり,低温で凍結されたマトリックス中と同様に3LLCTからの短波長発光が観測されたと考えた。一方,イオン交換量が増加するとC16TMAイオンの密度が疎となり層間の剛直性が失われ,室温下溶液中と同様にイリジウム錯イオンは3MLCTから発光したと考察した。実際,イオン交換量が多い試料に対し,77 Kで発光特性を観測すると,イオン交換量が少ないサンプルとほぼ同程度の短波長シフトが観測され,低温化による周囲環境の剛直化による影響が示された。このように,層間環境の剛直性を変化させることだけで発光特性を大きく変化させることができるのは,層間環境を自在に変化させることのできる二次元空間でのみ発現できる機能といえる。
また,二次元空間の剛直性を活かした短波長の高い発光エネルギーを溶液系へ取り出すため,テトラセン分子へのエネルギー移動について検討を行った。溶液中のイリジウム錯イオンは,室温では長波長発光しかしないため,テトラセン分子への振動の重なりがなく,共鳴エネルギー移動しない。一方,層状ケイ酸塩層間に導入した錯イオンは室温でも短波長発光するため,振動エネルギーの重なりが生じ,テトラセンへのエネルギー移動が観測された。この実験例は,ケイ酸骨格が創りだす空間を利用し,錯体周囲の環境を制御することで,錯体の光励起緩和過程を自在に操り,光エネルギーを取り出せることを意味している。
3. クリック反応を用いた交互積層金属酸化物による光誘起電荷分離制御
前項は,無機ホストとして,絶縁体であるケイ酸骨格を用い,ケイ酸骨格の創りだす空間を利用して,発光特性や電子移動などを制御した研究例を示した。本項では,無機ホストに酸化チタンのような半導体を用い,半導体ナノ構造を精緻に設計することで光励起電子移動を制御した研究を示す。
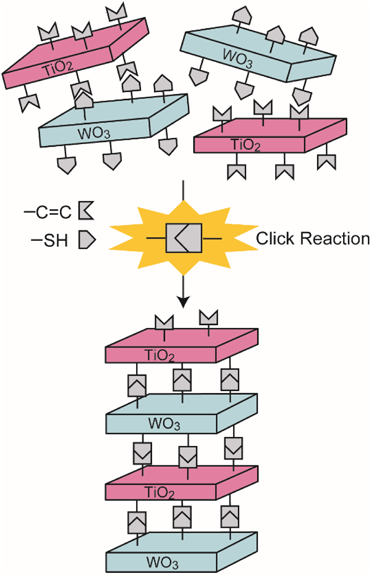
光励起による金属酸化物バンド構造の電荷分離状態の制御は,光触媒や人工光合成に重要である。電荷分離制御の一つとして,複数の金属酸化物種のナノメートルでの精密配置がある。我々は,クリック反応を利用した1ステップで交互積層構造を形成する手法を提案する(図5)12–14)。クリック反応は異種分子を選択的に結合させることができる反応の総称であり,有機高分子などの官能基化に近年広く利用されている。このクリック反応の一種であるチオール–エン反応は,チオール基とアルケン基が高収率に反応するため,官能基を導入したナノシートの交互積層体が形成する。さらに,本手法で得られる交互積層体は,層間に導入するアルキル鎖炭素数を変化させて交互積層体を合成することが可能となる。層間に導入したアルキル鎖炭素数を変化させ,層間距離を任意に制御した交互積層体を合成するとともに,得られた交互積層体の光触媒能や光励起電荷分離状態を評価する。
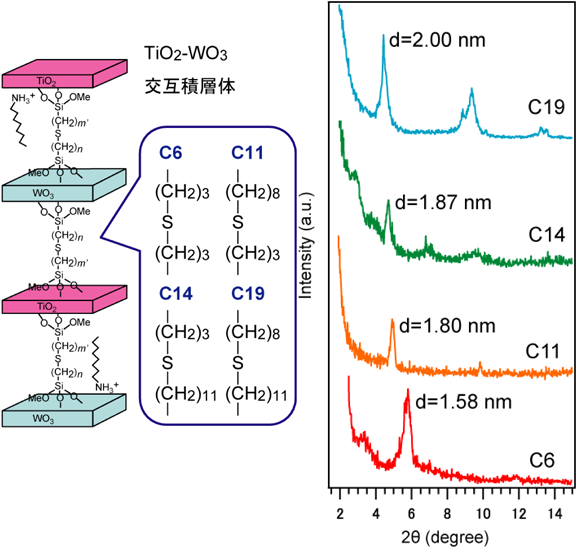
交互積層構造の合成は,層状チタン酸塩および層状タングステン酸塩を出発物質とした12)。層状チタン酸塩に対してシランカップリング剤を用いてアルケン基を,層状タングステン酸塩に対してシランカップリング剤を用いてチオール基を修飾した。それぞれの層状化合物をN,N-ジメチルホルムアミド中に分散して超音波処理を行うことで,チオール修飾チタン酸ナノシート分散液およびアルケン修飾タングステン酸ナノシート分散液を得た。2種のナノシートが等面積になるように分散液を混合し,ラジカル開始剤であるアゾビスイソブチロニトリルを加えることでチオール–エンクリック反応を進行させ,チタン酸ナノシートとタングステン酸ナノシートの交互積層体を得た。修飾するシランカップリング剤のアルキル鎖炭素数を変化させることで,交互積層体の層間距離を0.72,0.94,1.01,1.14 nmに制御することに成功した(図6)。
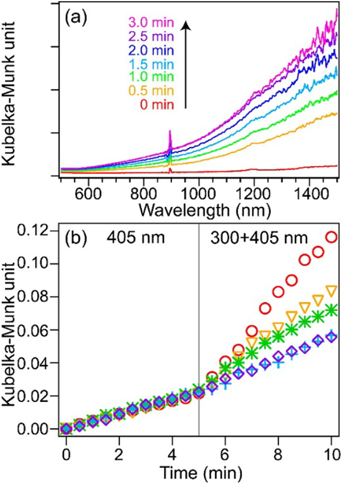
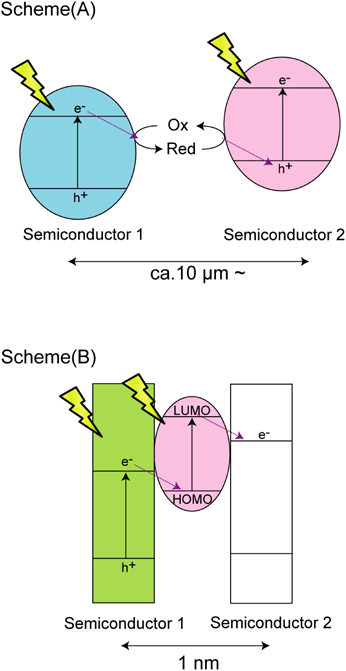
得られた交互積層体中にそれぞれの金属酸化物半導体のバンドギャップ幅以上のエネルギーを持つ紫外光を照射することで,チタン酸とタングステン酸の価電子帯電子は伝導帯に遷移する。チタン酸の伝導帯下端準位は,タングステン酸の伝導帯下端準位よりも負側(高いポテンシャルエネルギー)である。そのため,チタン酸の伝導帯電子はタングステン酸の伝導帯に移動し,電荷分離状態を形成することを予測し,この紫外光誘起電子移動反応が,交互積層体の層間距離に依存することを検証した。タングステン酸伝導帯電子は空気中の酸素を還元できず蓄積されるのに対し,チタン酸の伝導帯電子は空気中の酸素の還元によって消費されてしまう。更に,紫外光照射によって生じた価電子帯のホールは層間に存在する有機分子種によって消費され,タングステン酸伝導帯電子の再結合過程は抑制される。結果として,タングステン酸の伝導帯には,自己光励起および光誘起電子移動反応によって電子が蓄積される。我々は,この蓄積された電子量を近赤外拡散反射分光法によって定量的に観測することで,光誘起電子移動反応の評価を試みた(図7(a))。
交互積層体の粉末に対して,タングステン酸のみが吸収する405 nmの単色光を5 min間照射し,30 sごとに試料の近赤外領域の拡散反射スペクトルを測定した。この時の1000 nmにおけるクベルカムンク値は,光照射時間に対して増大した(図7(b))。このクベルカムンク値の増大は,タングステン酸の光励起による伝導帯電子量の増大に帰属できる。その後,300 nmと405 nmの単色光を同時に5 min間照射した。このとき,1000 nmにおけるクベルカムンク値の時間プロットの傾きは増大した。この傾きはチタン酸とタングステン酸の物理混合のものよりも大きかったことから,層間におけるチタン酸伝導帯からタングステン酸伝導帯への光誘起電子移動反応が起こっていることを支持する結果である。更に,層間距離の狭い交互積層体ほど,クベルカムンク値の傾きは大きい。したがって,層間距離が狭いほど,チタン酸からタングステン酸への光誘起電子移動反応が効率的に起こると結論することができる。
交互積層体の層間距離に依存した光触媒能を確認するために,この交互積層体の層間にメチレンブルーをイオン交換法によって導入し,メチレンブルーの光分解反応を行った。メチレンブルーが層間導入された交互積層体に300 nmと405 nmの光を照射し,メチレンブルーの分解量をUV-Vis吸収スペクトルから見積もった。層間距離の短い交互積層体ほど,より効率的に層間のメチレンブルーを分解することが確認できた。この結果は,交互積層体のナノ精密空間における光電荷分離過程を制御することで,交互積層体の光触媒能を向上させることが可能であることを示す。
交互積層体の層間でチタン酸からタングステン酸への紫外光誘起電子移動反応が起こることを示した(図8(a))。交互積層体の層間に色素分子を導入することで,可視光照射によって色素を介したタングステン酸からチタン酸への電子移動反応が起こることを期待した(図8(b))。実際に,交互積層体の層間にローダミンBをカチオン交換法によって導入し,可視光を照射することで,タングステン酸とローダミンBが光励起し,タングステン酸の伝導帯電子は光励起状態のローダミンBの空のHOMOレベルに移動し,光励起状態のローダミンBはチタン酸の伝導帯に移動することを検証した。光照射によって生成したタングステン酸の伝導帯電子は,ローダミンBの光励起によって効率的に消費されることを,近赤外拡散反射分光法によって示した。色素からチタン酸伝導帯準位への電子移動反応の実証は達成されていないが,この電子移動反応は典型的な色素増感反応であるため,容易に起こると期待している。
我々は,無機酸化物が提供するナノ空間・ナノ構造を自在に制御することで,光励起緩和過程を操る手法を開発してきた。ナノ材料を自在に制御する方法は,近年積極的に研究開発が行われ,ナノ空間・構造内にイオン・分子・分子集合体を配列することができるようになってきている。これらの手法を取り入れることにより,より効率的・選択的に光励起緩和過程を操作することができると期待される。
また,異種の材料を選択的に配列させることのできるクリック反応を利用したナノ材料設計手法は,その組み合わせの多様性から様々な次元の化合物との組み合わせを検討することにより,物質拡散などを二種界面で制御することで光化学応用以外にも触媒や電気化学などでの新たな展開が開けると考えている。