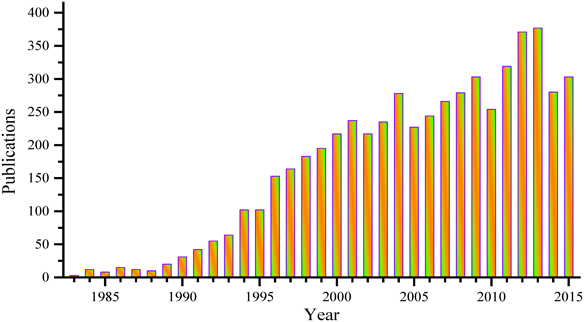
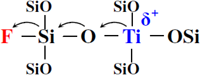30数年前にENI社は,MFI型ゼオライトの結晶骨格中への四配位Tiイオンの同形置換に初めて成功した1,2)。いわゆる第一世代のチタノシリケートTS-1を見出すことにより,石油精製,石油化学,ファインケミカルズ合成におけるゼオライトの応用を従来の固体酸触媒反応から液相選択酸化へ導いた。TS-1と過酸化水素から構成される触媒反応系はフェノールから二価フェノールへの水酸化,シクロヘキサノンのオキシム化3)またはプロピレンから酸化プロピレン(PO)への直接酸化(HPPOプロセス)4)において相次ぎ大規模に実用化され,多孔質材料化学と触媒化学にインパクトのある業績をもたらした。Fig. 1に示すように,それ以降も新規チタノシリケート材料と触媒作用に関する研究が盛んに行われ,数多くの報告がされている。
TS-1触媒が高い酸化活性を示す要因はMFI結晶構造の高い安定性,水の共存する液相中での適度な疎水性とユニークな三次元細孔構造に由来する2)。一方,その酸素10員環(MR)細孔は入り口が比較的に狭く顕著な形状選択性を示し,反応分子の細孔内への拡散とTi活性点へのアクセスを制限し,嵩高い基質酸化へのTS-1触媒の適用を妨げる。さらに,目標含酸素化合物の選択性についてもTS-1の特異な溶媒効果の影響で必ずしも実用化に満足できるとは言いがたい。また,異なるケイ素源,Ti源または有機型剤を用いるTS-1の製法が多数報告されたが,真に実用化に有効なのは,高価な有機ケイ素とTi源,テトラプロピルアンモニウムヒドロキシド(TPAOH)で水熱合成されたナノサイズ結晶であるとされる。TS-1におけるこれらの問題点を解消するため,細孔が酸素8員環から14員環を有する結晶性のチタノシリケートまたはTiを含有するメソポーラスシリカが相次ぎ直接水熱合成,ポスト合成,フッ化物法とドライゲル法で調製され,液相選択酸化触媒作用の多様化につながった(Table 1)。新規チタノシリケートにおいて,特にTi-MWW,Ti-MOR,Ti-MCM-68と層状TS-1が優れた触媒活性と生成物選択性を示し,実用化への触媒技術開発が期待されている。本稿では,紙面の制約で高性能層状チタノシリケートの合成と選択酸化触媒反応の特性に関する最近の基礎研究と工業化の結果を中心に解説する。
Table 1. A summary of titanosilicates.| Year | Material | FTC | Channels (MR) | Methoda | Ref. |
| 1983 | TS-1 | MFI | 10×10 | HTS | [1, 5–7] |
| 1989 | ETS-10 | | | HTS | [8,9] |
| 1990 | ETS-4 | | | HTS | [10] |
| 1990 | TS-2 | MEL | 10 | HTS | [11–13] |
| 1992 | Ti-Al-Beta | *BEA | 12×12 | HTS | [14, 15] |
| 1992 | Ti-ZSM-48 | *MRE | 10 | HTS | [16, 17] |
| 1994 | Ti-SAPO-5 | AFI | 12 | HTS | [18–20] |
| 1995 | Ti-ZSM-12 | MTW | 12 | HTS | [21] |
| 1995 | Ti-UTD-1 | DON | 14 | HTS | [22] |
| 1995 | Ti-UTD-8 | | | HTS | [22] |
| 1996 | Ti-Beta | *BEA | 12×12 | HTS, F-, DGC | [23–25] |
| 1996 | Ti-MOR | MOR | 12–8 | PS | [26,27] |
| 1998 | Ti-FER | FER | 10×8 | HTS | [28] |
| 1999 | Ti-ITQ-2 | MWWb | | PS | [29] |
| 2000 | Ti-MWW | MWW | 10–10 | HTS, PS | [30–33] |
| 2000 | Ti-ITQ-6 | FERc | | HTS+PS | [34] |
| 2000 | Ti-ITQ-7 | ISV | 12×12 | HTS | [35, 36] |
| 2004 | Ti-YNU-1 | MWWd | | HTS+PS | [37, 38] |
| 2007 | Ti-MCM-36 | MWWe | | HTS+PS | [39, 40] |
| 2008 | IEZ-Ti-MWW | MWWd | | HTS+PS | [41–43] |
| 2008 | Ti-MCM-56 | MWWf | | HTS+PS | [44] |
| 2008 | Ti-MCM-68 | MSE | 12×10×10 | PS | [45] |
| 2011 | L-TS-1 | MFI g | | HTS | [46, 47] |
| 2014 | Ti-ECNU-8 | FER | | PS | [48] |
| 1994 | Ti-MCM-41 | | 1D hexagonal | HTS | [49, 50] |
| 1994 | Ti-HMS | | 1D hexagonal | HTS | [51, 52] |
| 1995 | Ti-MSU | | | HTS | [53, 54] |
| 1996 | Ti-MCM-48 | | 3D cubic | HTS | [55–57] |
| 1999 | Ti-SBA-15 | | 1D hexagonal | HTS, PS | [57–61] |
| 2001 | Ti-MMS | | | HTS | [62] |
| 2002 | MTS-9 | | 1D hexagonal | HTS | [63, 64] |
| 2004 | Ti-MMM-1 | | | HTS | [65] |
| 2004 | Ti-MMM-2 | | | HTS | [66] |
| a HTS, hydrothermal synthesis; DGC, dry gel conversion; PS, post-synthesis; F−, fluoride media method. b Delaminated MWW (Del-MWW). c Delaminated FER (Del-FER). d Interlayer expanded MWW (IEZ-MWW). e Pillared MWW. f Partial delaminated MWW. g Lamellar MFI (L-MFI). |
2. 層状MFI型チタノシリケート(LTS-1)とTS-1微粒子の調製
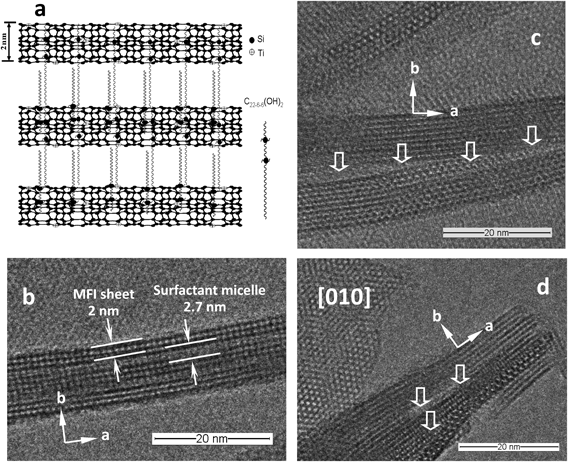
TPAOHを構造規定剤(structure-directing agent, SDA)に用いるTS-1の水熱合成系ではナノ粒子をさらに小さくして細孔内での拡散律速を緩和するには限界がある。Ryooらの研究グループは初めてミクロ構造とメソ構造を規定できるgeminiタイプ四級アンモニウム界面活性剤分子を設計し,この新しいタイプのSDAを用いて,本来,三次元結晶構造しかえられないZSM-5(MFI)ゼオライトのナノシートの水熱合成に成功した67)。この方法をTS-1の合成に適用し,親水的な四級アンモニウムカチオンがMFI構造のac面の結晶成長を促し,疎水的な炭化水素鎖が[010]面またはb軸方向の成長を抑制することにより,1単位ユニット厚さのナノシートがb軸方向に配列する層状TS-1(LTS-1)が合成された46,47)。LTS-1は焼成すると層間脱水縮合のミスマッチが発生し,結晶内にミクロ孔とメソ孔を共有する多孔物質または“card house”構造になる(Fig. 2)。この層状TS-1触媒は,通常のナノ粒子TS-1に比べ,鎖状アルケンのH2O2によるエポキシ化の活性は劣るが,メソ孔の存在によって分子サイズの大きい有機過酸化剤(TBHP)による環状アルケンまたはプロピレンのエポキシ化にはより有効で,嵩高い反応に高性能であった。
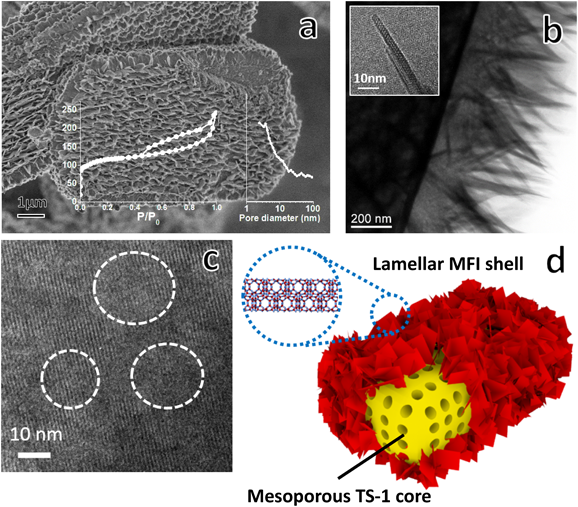
さらに,このような二元機能性SDAは,安価な方法で合成したTS-1のミクロンサイズの結晶のポスト処理に適用した場合,その触媒活性を大きく向上できる68)。あらかじめNaOH水溶液によってミクロンサイズのTS-1(8–12 µm)を脱ケイ素処理し,さらにgeminiタイプSDAで再結晶化を行うと,バルク状結晶表面にケイ素リッチな層状MFIゼオライトが形成するcore/shell構造が得られた(Fig. 3)。Coreのバルク結晶に本来のミクロ細孔以外に脱ケイ素によるメソ孔が生じた。Shell部分の層状構造にもメソ孔とミクロ孔が共存するため,得たもの(HTS-1)は階層の細孔構造を有する複合型多孔性物質である。バルク結晶TS-1はC3–C8の鎖状アルケンのH2O2によるエポキシ化に対して典型的なナノ結晶TS-1の30–50%程度の転化率しかないが,結晶表面に層状MFI構造を形成することにより,ナノ結晶TS-1の触媒活性と比較して約20–50%も増加した。層状shell部分はTiをほとんど持たないため,反応場または触媒活性点として直接に酸化反応に関与することはないと思われる。やはり階層のメソ孔の形成が結晶表面と細孔内への反応分子の濃縮,拡散または細孔に位置するTi活性点へのアクセスに有利に働くと推測される。この修飾法は生成物との分離が容易である一方,触媒機能が満足ではないミクロ結晶TS-1の有効利用につながる応用技術として使いうる。
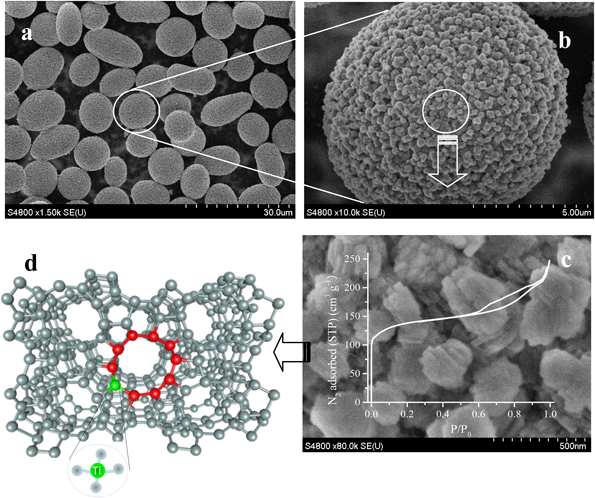
ケトンのオキシム化などの工業プロセスにおいて回分スラリー反応器を用いると分離し易いTS-1微粒子(microsphere)触媒が実際に多く使用さている。TS-1微粒子は水熱合成のナノ結晶を原料とし噴霧乾燥法(spray dry)により多工程を経て調製する方法が主流である。ミクロサイズの微粒子が直接水熱合成できれば,ゼオライトの調製過程ならびに反応プロセスでの分離にかかる手間が減らせる。TPAOHをSDAとするTS-1の合成系にP123とF127などのトリブロック共重合体(triblock copolymer)を共存させると,TS-1ナノ結晶が自己集合し5–15 µmの大きさのミクロ粒子を形成する(Fig. 4)69)。この球状粒子はTS-1ナノ結晶本来の物理化学性質と固有の酸化特性を保持しながら,一次粒子間に分子の拡散に有利なメソ孔も持つ。この球状触媒は,シクロヘキサノンの連続オキシム化において150時間反応を行っても結晶構造および球状構造が崩れることなく,実用化に応用できる強い機械強度を持つことがわかった70)。
3.1 調製法
MWW型ゼオライトは酸素12MRサイドポケットと互いに独立したジグザグな酸素10MR細孔と酸素12MRスーパーケージを含んだ10MR細孔を同時に有するユニークな構造を持つ71)。その三次元結晶構造は水熱合成において得られた層状前駆体の層間脱水縮合で形成されるため,代表的な層状ゼオライトともいえる。MWW構造へのTiの直接導入によるTi-MWWの調製は困難であったが,大量のホウ酸(Si/Bモル比0.75)を結晶化助剤とする水熱合成系でピペリジン(PI)またはヘキサメチレンイミン(HMI)をSDAに用いることにより,その層状前駆体の結晶化が可能になった30,72)。このように水熱合成したTi-MWWを真に有効な酸化触媒にするには,骨格外チタン種と一部の骨格ホウ素を選択的に酸処理によって除去することが不可欠であることもわかった。その後,脱ホウ素MWWシリケート,Ti源とPIの水溶液中で,三次元MWW構造を可逆的に二次元層状構造へ転換しながら骨格中にチタンを挿入するポスト法も開発された31)。この特殊なポスト法で調製したTi-MWW-PSは骨格中にチタンの触媒作用を妨害しうるホウ素をほとんど含まず,従来のホウ素を含有するTi-MWWに比べ直鎖アルケンのエポキシ化反応において高い酸化活性を示す。これらの結果は本誌での解説73)または成書74)を参考にされたい。
Ti-MWWの直接合成でホウ素添加量を減らす方法もいくつか見出された。dry-gel conversion(DGC)法を用いることで,Ti-MWWの直接水熱合成に必要なホウ素量を大きく減らすことができた32)。また,dry-gelに脱ホウ素MWWシリケートを種結晶として10%加え,Si/Bモル比12の条件下でもTi-MWWを結晶化できる。しかし,DGC法のTi-MWWは結晶粒子が水熱法の10–20倍も大きくなるため,エポキシ化活性は水熱合成のTi-MWW触媒にははるかに及ばない。一方,従来のホウ素を含む水熱合成系にフッ化水素酸を添加するフッ化物法でもホウ素の使用量を大幅に減らせる。HF/Si=1,HMI/Si=1の条件下でSi/B=6の合成ゲルからもTi-MWWが十分に結晶化できる75)。フッ化物法で得たTi-MMW-Fは結晶サイズが通常水熱法のものより大きくなるが,1-ヘキセンのエポキシ化において同程度かむしろやや高い触媒活性を示す。フッ化物法による触媒粒子の疎水性の向上またはTi活性点のフッ素による化学的な修飾が触媒の活性向上に寄与すると推測される。また,Ti-MWW-Fの層状前駆物はポスト処理における構造変化の特異性も示す。その層状前駆体を酸処理しさらに焼成すると,本来ならば2D層状構造が3D MWW結晶構造へ転換するはずであるが,しばしば層間に珪素が挿入される拡張構造をとる。これは,フッ化物法で得られる結晶が層に平行となる結晶面が特別により長くなり,ケイ素などのゲスト物による層間のピラーリングに有効に働くためと思われる。
PIやHMIなどの環状アミンは合成手法が異なっても,Ti-MWW合成用の典型的なSDAである。しかし,オクチル基またはヘプチル基などの置換基をもつ直鎖アルキルトリメチルアンモニウムヒドロキシド(OCTMAOH, HEPTMAOH)をSDAに用いると,同時に適度なカリウムイオン(K/Si=0.04–0.06)が鉱化剤として存在する条件下でホウ酸助剤の少ないゲル(Si/B=10)からTi-MMWを直熱水熱合成できる76,77)。この合成系でMWW構造を結晶化できるTiの添加量の範囲は比較的狭い(Si/Ti=40–100)。また,MFI或いはアモルファスが生成しやすいため,ゲル調製と結晶化過程の精密な制御が不可欠である。SDAのサイズをさらに短いヘキシルトリメチルアンモニウム(HEXTMAOH)にすると,MFI構造しか合成できない。
ホウ酸結晶化助剤またはHF鉱化剤を全く使用せずTi-MWWを直接水熱合成できるならば,最も理想的である。MWW構造の特徴に基づき,層内の10MR細孔と層間の10MR細孔にそれぞれ進入できる分子サイズの異なるSDAを用い,結晶化の段階で構造安定化と空間充填を同時に実現させる方法,いわゆる“dual SDA”法によってホウ酸を含まない条件下でTi-MWWの調製が可能となった33)。1-アダマンチルトリメチルアンモニウムヒドロキシド(TMAadOH)とHMIが共存するゲルから純シリカMWW,ITQ-1を合成することはできるが,AlやTiなどのヘテロイオンを加えるとMWW構造の結晶化が進行しなくなるとされている78–81)。しかし,少量のカリウム(K/Si=0.07)鉱化剤を加えるとホウ酸を含まない条件下でTi-MWWを水熱合成できることがはじめてわかった。得られたTi-MWW-Dualは結晶化過程でTiがホウ素の妨害を受けずに骨格に自由に導入できるため,先述のポスト法Ti-MWW-PSと同様にホウ酸助剤法で合成したTi-MWW-HTSと比べアルケンのエポキシ化においてより高い酸化活性を示す。Table 2にこれまで開発されたTi-MWWの製法と特徴をまとめる。
Table 2. A summary of preparation methods for Ti-MWW.| Methoda | Gel composition | SDA | Ti-MWW product | Ref. |
| Si/Ti ratio | Si/B ratio | crystal size | catalytic activity b |
| HTS | 20–100 | 0.75 | PI, HMI | nanosize | 150–250 | [30, 72] |
| PS | 20–100 | >100 | PI, HMI | nanosize | 300–450 | [31] |
| DGC | 50–100 | 1.0–12 | PI, HMI | microsize | <120 | [32] |
| F− | 20–100 | 6 | HMI | microsize | 200–350 | [75] |
| HTS | 40–100 | 5–20 | OCTMAOH, HEPTMAOH | nanosize | 200–300 | [76, 77] |
| Dual HTS | 30–100 | ∞ | TMAadOH+HMI | nanosize | 250–450 | [33] |
| a HTS, hydrothermal synthesis; DGC, dry gel conversion; PS, post-synthesis; F−, fluoride media method. b Expressed in turnover number (TON) per Ti site for alkene epoxidation: 1-hexene or H2O2 (30%), 10 mmol; MeCN solvent, 10 mL; temp., 333 K; time, 2 h. |
3.2 ポスト処理による構造修飾
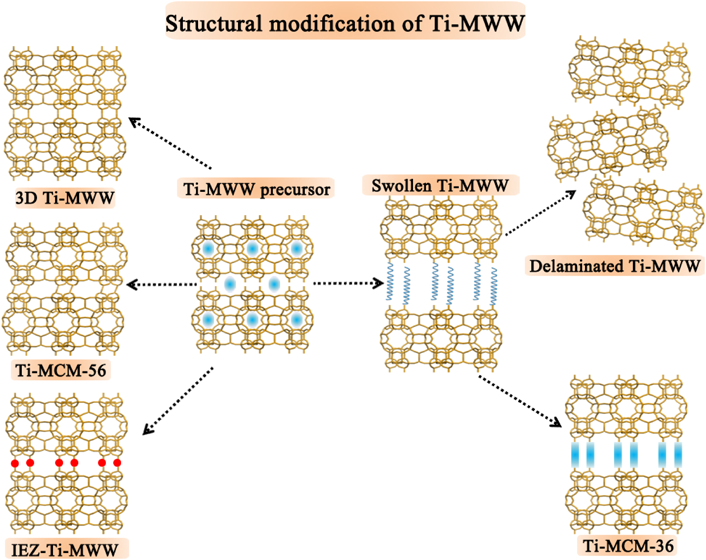
層状物質であるTi-MWWは結晶構造に柔軟性,多様性,可塑性をあわせ持つ。層状前駆体に対し層間膨張,剥離,ピラー化および層間拡張などのポスト処理で広い反応場を有する構造改良型Ti-MWW触媒が得られる(Fig. 5)。塩基性条件下において臭化セチルトリメチルアンモニウム(CTAB)界面活性剤で層間構造を膨張させてから超音波処理を行うと,高い外表面積を持ち分子サイズの大きなアルケンのエポキシ化に有効な完全層剥離Del-Ti-MWW触媒が調製できる82,83)。一方,界面活性剤で膨張したものについて層間にシリカなどのピラーを挿入し,上下のMWWナノシートを支えることにより層間にメソ空間をつくることで,ミクロ孔とメソ孔を共有するハイブリッドチタノシリケートTi-MCM-36触媒が得られる39)。
膨張と超音波処理を組み合わせる完全剥離に対し,より温和な酸処理でMWWナノシートの配列を無造作にずらすことで層間を部分的に剥離することが可能である。Ti-MWWの層状前駆体を低濃度の硝酸を用いて室温で短時間処理すると,層間に位置する有機物が部分的に取り除かれ,ナノシートの規則的な配列が乱されることで,構造が直接水熱合成のMCM-56に非常によく似た類縁物Ti-MCM-56が得られる44)。さらに,Ti-MCM-56を焼成しても部分的な層間剥離構造は保たれる。得られた材料はSub-Zeoliteとも称される84)。3D結晶構造のTi-MWWと比べ,Ti-MCM-56はより広い外表面に露出している活性点を持ち,数多くのバルキーな分子の選択酸化反応に優れた活性を示す44)。この簡単かつコントロールし易いポスト法はFER構造とNSI構造の層状前駆体の層間剥離にも適応できる48,85)。
PIまたはHMIのポスト処理を施して3D MWWと2D層状物の可逆的な3D↔2D構造転換を利用して得たTi-MWW層状前駆体は,酸処理と焼成の後でも層間が完全に縮合せず拡張を保ったTi-YNU-1が偶然に見つけられた37)。Ti-YNU-1は通常の三次元構造を有する3D Ti-MWWより層間距離が約0.25 nm広くなり,より広い入り口を層間に持つ。後の構造解析により酸処理で洗い出されたケイ素が層間に挿入され層間拡張構造に関与すると結論づけられた86)。この知見から層間に化学組成と構造の明確なシラン修飾剤を挿入し,新規な三次元結晶構造を創生する汎用的な技術が開発された。得たものを層間拡張ゼオライト(Interlayer-Expanded Zeolite, IEZ)と名づける41)。Ti-MWW層状前駆体に対して層間にモノシラン剤を直接挿入すると12MR構造のIEZ-Ti-MWWが得られる41,42)。一方,CTABで層間拡張した前駆体を二量体型シラン剤でシリル化処理すると,層間細孔の員環数が14MRになるものを調製できる43)。
総括的にFig. 5に示した構造修飾の手法は,Ti-MWWの酸化触媒活性を向上させるだけではなく広い反応場を必要とする大分子の触媒作用にも役立つ。
3.3 チタン活性点の化学修飾によるTi-MWW触媒作用の促進
構造修飾は主に反応分子の拡散と活性点へのアクセスを有利にさせる観点からの触媒の改良方法である。それ以外にゼオライト骨格の物理化学性質ならびにそこに位置する活性点のミクロスコピックな化学的環境を改善することも触媒活性の向上につながると考えられる。
前述のPIまたはHMI処理で3D Ti-MWWをいったん可逆的2D層状物質に反転し,さらに焼成で再び3D構造に転換すると,Tiの含有量と配位状態が変化せず,全構造の再構成による欠陥サイトが約35%減少し,Ti-MWWの疎水性の増加,またはエポキシ化活性の向上につながった87,88)。この方法が失活したTi-MWW触媒の再生に効果的であることもわかっている89)。
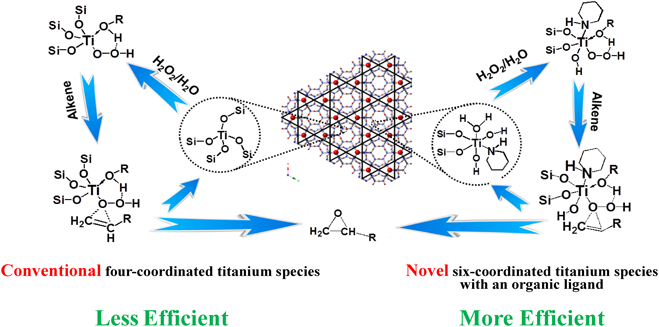
最近,PI処理による構造転換で得た有機アミン分子を含有する2D構造のTi-MWW-PIも比表面積が低いにもかかわらず非常に高活性な触媒であることが見出された90)。実験評価と理論計算から,PI分子が配位子としてTi-MWW-PIに六配位チタン活性種を与えると考えられる(Fig. 6)。このようなopen Ti種は3D Ti-MWWの通常のclosed Ti種より高いH2O2活性化能力を持ち,1-ヘキセンのエポキシ化において2倍以上のTONを示す。
それ以外にも,Ti-MWW層状前駆体を酸処理する過程で,適当なNH4Fを添加することにより,ゼオライト骨格にフッ素を導入することができる。固体19F MAS NMR測定から(SiF6)2−,(SiO2/4F)−とSiO2/3Fなどのフッ素種が形成されることがわかった91,92)。骨格フッ素がゼオライトの疎水性を増加するだけではなく,Fig. 7に示すSiO2/3F種がフッ素の強い求電子作用によりTi活性点とH2O2分子の化学的な作用を強めることで,フッ素化したTi-MWW-Fは通常のTi-MWWに比べ高いエポキシ化活性を示す。
チタノシリケートは過酸化水素を酸化剤とする液相酸化の触媒として,主にアルケン(官能基を持つ化合物も含む)のエポキシ化,ケトンとアルデヒド類のオキシム化,芳香族(フェノールとベンゼンなど)の水酸化に有効である。TS-1はこの三種類の反応に最も有効なチタノシリケート触媒であったが,Ti-MWW,Ti-MOR,Ti-MCM-68などの新規触媒が発見されるにつれ状況が変わりつつある。Ti-MWWの触媒作用を詳しく検討した結果,芳香族の水酸化には有効ではないが,エポキシ化とオキシム化においては優れた特性を示すことがわかった。エチレン93),プロピレン94),塩化アリル89),アリルアルコール95),ジアリルエーテル96)と2,5-ジヒドロフラン97,98)のエポキシ化に対して高い転化率と過酸化水素の有効利用率を示すのみならず,特に高いエポキシド選択性(多くの場合,>99%)を有する。これはエポキシ化におけるTi-MWWとTS-1の溶媒効果が全く異なるからである。プロトン性のメタノールがTS-1に有利であるに対し,Ti-MWWは,エポキシドとの開環反応に不活性でかつ塩基性を有するアセトニトリル溶媒中において最大の触媒作用を発揮する99)。溶媒効果の相違は,各々のゼオライトの結晶骨格と細孔の構造,親水性・疎水性にかかわるとされるが,原因究明にはより深い研究が必要である。しかし,結果として不活性なアセトニトリルを好むTi-MWWがPOなどのバルクケミカルの製造において選択性の高いプロセスを作り出すことが期待できる。実際にTS-1/H2O2によるHPPO実用プロセスでは,メタノール溶媒によるPOの開環反応が避けられず,高沸点副生成物の生成による高い分離コストと触媒の急速な失活などの問題点を抱えている。
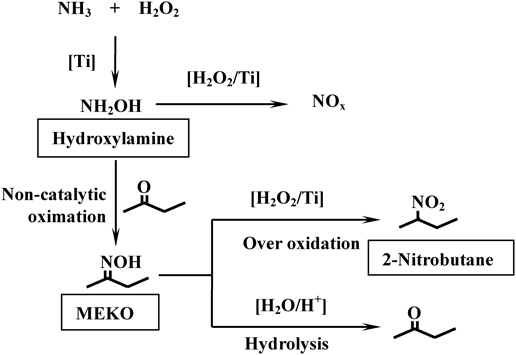
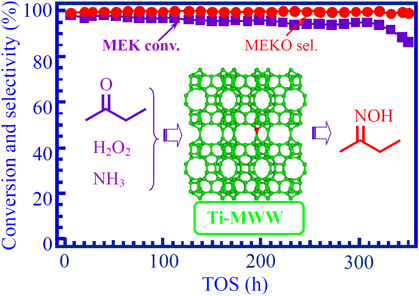
オキシム化においてTi-MWW触媒は,水またはターシャリーブチルアルコール溶媒中においてシクロヘキサノン87,100)とメチルエチルケトン(MEK)の反応101)に非常に有効で,TS-1より優れた触媒作用を示す。特に鎖状ケトンMEKのオキシム化ではTS-1に見られないメチルエチルケトンオキシム(MEKO)への選択性(>99%)を示す。TS-1触媒の場合,MEKOのH2O2による過剰酸化を防げず,最大20%の2-ニトロブタンを副生してしまう。反応メカニズムを検討したところ,ケトンのオキシム化はチタン活性点上でアンモニアがH2O2に酸化されヒドロキシルアミンを生成し,さらにこの中間体とケトン分子との無触媒反応でオキシムを生成する(Fig. 8)。チタノシリケートは主反応の進行に重要なだけではなく,触媒劣化の要因であるヒドロキシルアミン中間体の酸化分解またはオキシム生成物の過剰酸化にも密接にかかわる。オキシム化は過剰なアンモニアが存在する塩基性条件下で進行するため,触媒の失活は細孔内への高沸点有機物の吸着のほか,ゼオライト骨格の脱ケイ素による溶解または構造崩壊が主因である。それを完全に阻止することはできないが,反応系に適度のシリカを添加すること87),またはTi-MWWの結晶をメソポーラスシリカで被覆するcore/shell構造を構築することで,シリカが脱ケイ素の犠牲剤として働き,オキシム化でのTi-MWW触媒寿命を引き伸ばすことができる102)。
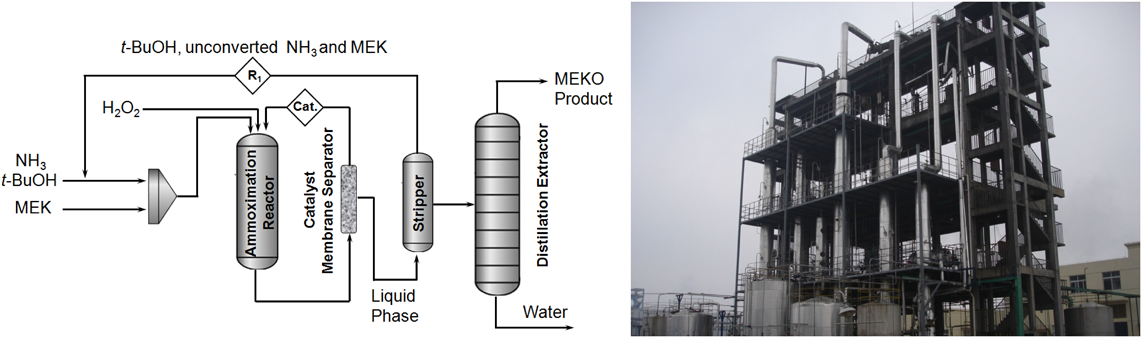
Ti-MWW材料の合成とその触媒作用の基礎研究に基づき触媒調製とMEKO選択合成プロセスの工業化技術が得られた。工業化Ti-MWW触媒は,MEKケトンの連続オキシム化において長い寿命と高いMEKO選択性を維持することができる(Fig. 9)103)。これらの触媒技術からオキシム化,触媒の膜分離,生成物分離の三つの主工程から構成されるクリーンなMEKO製造プロセス(1.5万トン/年)が開発され,中国で実用化された。従来の硫酸ヒドロキシルアミン法では大量の硫酸アンモニウムと汚水が排出されるに対し,新規環境調和型プロセスは水のみが副生成物である。
TS-1の発見以来,チタノシリケートの合成法,活性点の構造解析と修飾法また触媒作用機構に関する研究で確立された知見が選択酸化反応の大規模な工業化学プロセスへの応用に貢献した。そのうち,層状Ti-MWWに関する基礎研究と応用技術の成果は新しいオキシム化プロセスの工業化に発展し,今後より画期的なHPPO製造プロセスへの応用も期待できる。本稿では層状TS-1とTi-MWWチタノシリケートを中心に研究進展状況を紹介したが,SDAを使用しないポスト法で調製されたTi-MCM-68とTi-MORの触媒作用に関する研究も盛んに行われている。Ti-MCM-68はTS-1などに見られない高いフェノール水酸化活性とパラ選択性を持ち45,104),よりクリーンな芳香族水酸化反応への応用が期待される。また,ケトンとアルデヒドのオキシム化に有効なTi-MORについてはすでにMEKOとシクロヘキサノンオキシムの製造への工業応用が現実味を帯びている105–110)。ポスト法で調製されるチタノシリケート触媒も実用化に役立つことが実証されるだろう。さらに,チタノシリケートと貴金属Au, Pdの二元機能触媒でH2O2の代わりにH2とO2を用い,プロピレンのPOへの選択酸化が可能になった111–113)。また,チタノシリケートとRh(OH)xを組み合わせ,オキシム化と芳香属アルデヒドオキシム異性化を触媒作用しアミド類をone-pot合成する触媒系も開発された114,115)。これらの研究の詳細は紙面の制限で今回割愛さていただきたい。
謝辞Acknowledgments
本研究に当たって,中国華東師範大学何鳴元教授,劉月明教授,東京工業大学辰巳敬名誉教授(現NITE),八嶋建明名誉教授,小松隆之教授のご指導・ご協力をいただいたことをここに表記し,心より謝意を申し上げます。
引用文献References
1) M. Taramasso, G. Perego, and B. Notari, U.S. Pat., 4,410,501 (1983).
2) G. Bellussi, and M. S. Rigutto, Stud. Surf. Sci. Catal., 137, 911 (2001).
3) P. Roffia, G. Leofanti, A. Cesana, M. Mantegazza, M. Padovan, G. Petrini, S. Tonti, and P. Gervasutti, Stud. Surf. Sci. Catal., 55, 43 (1990).
4) D. H. Wells Jr., W. N. Delgass Jr., and K. T. Thomson, J. Am. Chem. Soc., 126, 2956 (2004).
5) Y. K. Hwang, J. S. Chang, S. E. Park, D. S. Kim, Y. U. Kwon, S. H. Jhung, J. S. Hwang, and M. S. Park, Angew. Chem. Int. Ed., 44, 556 (2005).
6) C. H. Xu, T. Jin, S. H. Jhung, J. S. Chang, J. S. Hwang, and S. E. Park, Catal. Today, 111, 366 (2006).
7) W. Fan, R. G. Duan, T. Yokoi, P. Wu, Y. Kubota, and T. Tatsumi, J. Am. Chem. Soc., 130, 10150 (2008).
8) S. M. Kuznicki, U.S. Pat., 4,853,202 (1989).
9) X. Yang, J. L. Paillaud, H. F. W. J. van Breukelen, H. Kessler, and E. Duprey, Micropor. Mesopor. Mater., 46, 1 (2001).
10) S. M. Kuznicki, U.S. Pat., 4,938,939 (1990).
11) J. S. Reddy, R. Kumar, and P. Ratnasamy, Appl. Catal., 58, L1 (1990).
12) J. S. Reddy, and S. Sivasanker, Catal. Lett., 11, 241 (1991).
13) J. S. Reddy, and R. Kumar, Zeolites, 12, 95 (1992).
14) M. A. Camblor, A. Corma, A. Martinez, and J. Perez-Pariente, J. Chem. Soc., Chem. Commun., 589 (1992).
15) Y. Goa, P. Wu, and T. Tatsumi, J. Phys. Chem. B, 108, 8401 (2004).
16) D. P. Serrano, H-X. Li, and M. E. Davis, J. Chem. Soc., Chem. Commun., 745 (1992).
17) K. M. Reddy, S. Kaliaguine, and A. Sayari, Catal. Lett., 23, 175 (1994).
18) A. Tuel, and Y. B. Taârit, J. Chem. Soc., Chem. Commun., 1667 (1994).
19) A. Tuel, Zeolites, 15, 228 (1995).
20) B. Y. Hsu, S. Cheng, and J. M. Chen, J. Mole. Catal. A: Chem., 149, 7 (1999).
21) A. Tuel, Zeolites, 15, 236 (1995).
22) K. J. Balkus Jr, A. G. Gabrielov, and S. I. Zones, Stud. Surf. Sci. Catal., 97, 519 (1995).
23) M. A. Camblor, M. Costantini, A. Corma, L. Gilbert, P. Esteve, A. Martinez, and S. Valencia, Chem. Commun., 1339 (1996).
24) T. Blasco, M. A. Camblor, A. Corma, P. Esteve, A. Martinez, C. Prieto, and S. Valencia, Chem. Commun., 2367 (1996).
25) N. Jappar, Q. Xia, and T. Tatsumi, J. Catal., 180, 132 (1998).
26) P. Wu, T. Komatsu, and T. Yashima, J. Phys. Chem., 100, 10316 (1996).
27) P. Wu, T. Komatsu, and T. Yashima, Stud. Surf. Sci. Catal., 105, 663 (1997).
28) R. K. Ahdei, and A. N. Kotasthane, J. Mater. Chem., 8, 1685 (1998).
29) A. Corma, U. Diaz, V. Fornes, J. L. Jorda, M. Domine, and F. Rey, Chem. Commun., 779 (1999).
30) P. Wu, T. Tatsumi, T. Komatsu, and T. Yashima, Chem. Lett., 29, 774 (2000).
31) P. Wu, and T. Tatsumi, Chem. Commun., 1026 (2002).
32) P. Wu, T. Miyaji, Y. Liu, M. He, and T. Tatsumi, Catal. Today, 99, 233 (2005).
33) N. Liu, Y. Liu, W. Xie, L. Wang, M. He, and P. Wu, Stud. Surf. Sci. Catal., 170, 464 (2007).
34) A. Corma, U. Diaz, M. E. Domine, and V. Fornes, Angew. Chem. Int. Ed., 39, 1499 (2000).
35) M. J. Diaz-Cabanas, L. A. Villaescusa, and M. A. Camblor, Chem. Commun., 761 (2000).
36) A. Corma, M. J. Diaz-Cabanas, M. E. Domine, and F. Rey, Chem. Commun., 1725 (2000).
37) W. Fan, P. Wu, S. Namba, and T. Tatsumi, Angew. Chem. Int. Ed., 43, 236 (2004).
38) X. Shen, W. Fan, Y. He, P. Wu, J. Wang, and T. Tatsumi, Appl. Catal. A: General, 401, 37 (2011).
39) S. Y. Kim, H. J. Ban, and W. S. Ahn, Catal. Lett., 113, 160 (2007).
40) F. Jin, S. Y. Chen, L. Y. Jang, J. F. Lee, and S. Cheng, J. Catal., 319, 247 (2014).
41) P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki, and T. Tatsumi, J. Am. Chem. Soc., 130, 8178 (2008).
42) L. Wang, Y. Wang, Y. Liu, H. Wu, X. Li, M. He, and P. Wu, J. Mater. Chem., 19, 8594 (2009).
43) H. Xu, L. Fu, J-G. Jiang, M. He, and P. Wu, Micropor. Mesopor. Mater., 189, 41 (2014).
44) L. Wang, Y. Wang, Y. Liu, L. Chen, S. Cheng, G. Gao, M. He, and P. Wu, Micropor. Mesopor. Mater., 113, 435 (2008).
45) Y. Kubota, Y. Koyama, T. Yamada, S. Inagaki, and T. Tatsumi, Chem. Commun., 6224 (2008).
46) K. Na, C. Jo, J. Kim, W. S. Ahn, and R. Ryoo, ACS Catal., 1, 901 (2011).
47) J. Wang, L. Xu, K. Zhang, H. Peng, H. Wu, J. Jiang, Y. Liu, and P. Wu, J. Catal., 288, 16 (2012).
48) B. Yang, and P. Wu, Chin. Chem. Lett., 25, 1511 (2014).
49) A. Corma, M. T. Navarro, and J. P. Pariente, J. Chem. Soc., Chem. Commun., 147 (1994).
50) T. Blasco, A. Corma, M. T. Navarro, and J. P. Pariente, J. Catal., 156, 65 (1995).
51) P. T. Tanev, M. Chibwe, and T. J. Pinnavaia, Nature, 368, 321 (1994).
52) W. Zhang, J. Wang, P. T. Tanev, and T. J. Pinnnavaia, Chem. Commun., 979 (1996).
53) S. A. Bagshaw, E. Prouzet, and T. J. Pinnavaia, Science, 269, 1242 (1995).
54) P. Wu, H. Sugiyama, and T. Tatsumi, Stud. Surf. Sci. Catal., 146, 613 (2003).
55) K. A. Koyano, and T. Tatsum, Chem. Commun., 145 (1996).
56) M. Morey, A. Davidson, and G. Stucky, Micropor. Mater., 6, 99 (1996).
57) M. S. Morey, S. ƠBrien, S. Schwarz, and G. D. Stucky, Chem. Mater., 12, 898 (2000).
58) Z. Luan, E. M. Mase, P. A. W. van der Heide, D. Zhao, R. S. Czernuszewicz, and L. Kevan, Chem. Mater., 11, 3680 (1999).
59) B. L. Newalkar, J. Olanrewaju, and S. Komarneni, Chem. Mater., 13, 552 (2001).
60) P. Wu, T. Tatsumi, T. Komatsu, and T. Yashima, Chem. Mater., 14, 1657 (2002).
61) W. H. Zhang, J. Lu, B. Han, M. Li, J. Xiu, P. Ying, and C. Li, Chem. Mater., 14, 3413 (2002).
62) W. S. Ahn, N. K. Kim, and S. Y. Jeong, Catal. Today, 68, 83 (2001).
63) F. S. Xiao, Y. Han, Y. Yu, X. Meng, M. Yang, and S. Wu, J. Am. Chem. Soc., 124, 888 (2002).
64) F. S. Xiao, Y. Han, X. Meng, Y. Yu, M. Yang, and S. Wu, Stud. Surf. Sci. Catal., 146, 565 (2003).
65) P. Zhao, C. Yu, Z. Peng, and C. Xia, J. Mole. Catal. (China), 18, 208 (2004).
66) O. A. Kholdeeva, M. S. Mel’gunov, A. N. Shmakov, N. N. Trukhan, V. V. Kriventsov, V. I. Zaikovskii, M. E. Malyshev, and V. N. Romannikov, Catal. Today, 91–92, 205 (2004).
67) M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki, and R. Ryoo, Nature, 461, 246 (2009).
68) C. G. Li, Y. Lu, H. Wu, P. Wu, and M. He, Chem. Commun., 51, 14905 (2015).
69) Z. Wang, L. Xu, J. Jiang, Y. Liu, M. He, and P. Wu, Micropor. Mesopor. Mater., 156, 106 (2012).
70) Z. Wang, L. Xu, M. He, and P. Wu, Chem. Rea. Eng. Technol. (in Chinese), 29, 456 (2013).
71) M. E. Leonowicz, J. A. Lawton, S. L. Lawton, and M. K. Rubin, Science, 264, 1910 (1994).
72) P. Wu, T. Tatsumi, T. Komatsu, and T. Yashima, J. Phys. Chem. B, 105, 2897 (2001).
73) 呉 鵬,辰巳 敬,ゼオライト,20, 147 (2003).
74) P. Wu, H. Xu, L. Xu, Y. Liu, and M. He, “MWW-Type Titanosilicate-Synthesis, Structural Modification and Catalytic Applications”, SPRINGER BRIEFS IN MOLECULAR SCIENCE GREEN CHEMISTRY FOR SUSTAINABILITY, Springer, ISBN: 978–3–642–39114–9, 2013.
75) P. Wu, K. Sugimoto, and T. Tatsumi, unpublished results.
76) N. Liu, Y. Liu, C. Yue, L. Chen, H. Wu, X. Li, M. He, and P. Wu, Chem. Lett., 36, 916 (2007).
77) C. Yue, W. Xie, Y. Liu, H. Wu, X. Li, and P. Wu, Micropor. Mesopor. Mater., 142, 347 (2011).
78) M. J. Diaz Cabanas, M. A. Camblor Fernandez, C. Corell Matires, and A. Corma Camos, W.O. Pat., 9,719,021 (1997).
79) S. Nicolopoulos, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. A. Camblor, A. Corma, and M. J. Diaz-Cabanas, J. Am. Chem. Soc., 119, 11000 (1997).
80) M. A. Camblor, A. Corma, M-J. Diaz-Cabanas, and C. Baerlocher, J. Phys. Chem. B, 102, 44 (1998).
81) A. Corma, C. C. A. Catlow, and G. Sastre, J. Phys. Chem. B, 102, 7085 (1998).
82) N. Duangamol, P. Wu, and T. Tatsumi, Chem. Lett., 32, 326 (2003).
83) P. Wu, N. Duangamol, J. Ruan, Y. Liu, M. He, W. Fan, O. Terasaki, and Takashi Tatsumi, J. Phys. Chem. B, 108, 19126 (2004).
84) W. J. Roth, P. Nachtigall, R. E. Morris, and J. Čejka, Chem. Rev., 114, 4807 (2014).
85) H. Xu, L. Jia, H. Wu, B. Yang, and P. Wu, Dalton Trans., 43, 10492 (2014).
86) J. Ruan, P. Wu, B. Slater, and O. Terasaki, Angew. Chem. Int. Ed., 44, 6719 (2005).
87) F. Song, Y. Liu, H. Wu, M. He, P. Wu, and T. Tatsumi, J. Catal., 237, 359 (2006).
88) L. Wang, Y. Liu, W. Xie, M. He, and P. Wu, J. Phys. Chem. C, 112, 6132 (2008).
89) L. Wang, Y. Liu, W. Xie, H. Zhang, H. Wu, Y. Jiang, M. He, and P. Wu, J. Catal., 246, 205 (2007).
90) L. Xu, D. D. Huang, C. G. Li, X. Ji, S. Jin, Z. Feng, F. Xia, X. Li, F. Fan, C. Li, and P. Wu, Chem. Commun., 51, 9010 (2015).
91) X. Fang, Q. Wang, A. Zheng, Y. Liu, Y. Wang, X. Deng, H. Wu, F. Deng, M. He, and P. Wu, Catal. Sci. Technol., 2, 2433 (2012).
92) X. Fang, Q. Wang, A. Zheng, Y. Liu, L. Lin, H. Wu, F. Deng, M. He, and P. Wu, Phys. Chem. Chem. Phys., 15, 4930 (2013).
93) X. Lu, W-J. Zhou, H. Wu, A. Liebens, and P. Wu, Appl. Catal. A: General, 515, 51 (2016).
94) F. Song, Y. Liu, L. Wang, H. Zhang, M. He, and P. Wu, Stud. Surf. Sci. Catal., 170, 1236 (2007).
95) P. Wu, and T. Tatsumi, J. Catal., 214, 317 (2003).
96) P. Wu, Y. Liu, M. He, and T. Tatsumi, J. Catal., 228, 183 (2004).
97) H. Wu, L. Wang, H. Zhang, Y. Liu, P. Wu, and M. He, Green Chem., 8, 78 (2006).
98) H. Wu, Y. Liu, L. Wang, H. Zhang, M. He, and P. Wu, Appl. Catal. A: General, 320, 173 (2007).
99) W. Fan, P. Wu, and T. Tatsumi, J. Catal., 256, 62 (2008).
100) F. Song, Y. Liu, H. Wu, M. He, P. Wu, and T. Tatsumi, Chem. Lett., 34, 1436 (2005).
101) F. Song, Y. Liu, L. Wang, H. Zhang, M. He, and P. Wu, Appl. Catal. A: General, 327, 22 (2007).
102) L. Xu, H. Peng, K. Zhang, H. Wu, L. Chen, Y. Liu and P. Wu, ACS Catal., 3, 103 (2013).
103) S. Zhao, W. Xie, Y. Liu, and P. Wu, Chin. J. Catal., 32, 179 (2011).
104) S. Inagaki, Y. Tsuboi, M. Sasaki, K. Mamiya, S. Park, and Y. Kubota, Green Chem., 18, 735 (2016)
105) H. Xu, Y. Zhang, H. Wu, Y. Liu, X. Li, J. Jiang, M. He, and P. Wu, J. Catal., 281, 263 (2011).
106) L. Xu, J. Ding, Y. Yang, and P. Wu, J. Catal., 309, 1 (2014).
107) Y. Yang, J. Ding, B. Wang, J. Wu, C. Zhao, G. Gao, and P. Wu, J. Catal., 320, 160 (2014).
108) Y. Yang, J. Ding, C. Xu, W. Zhu, and P. Wu, J. Catal., 325, 101 (2015).
109) J. Ding, L. Xu, Y. Yu, H. Wu, S. Huang, Y. Yang, J. Wu, and P. Wu, Catal. Sci. Technol., 3, 2587 (2013).
110) J. Ding, and P. Wu, Appl. Catal. A: General, 488, 86 (2014).
111) L. Xu, Y. Ren, H. Wu, Y. Liu, Z. Wang, Y. Zhang, J. Xu, H. Peng, and P. Wu, J. Mater. Chem., 21, 10852 (2011).
112) Y. Ren, L. Xu, L. Zhang, J. Wang, Y. Liu, M. He, and P. Wu, Pure Appl. Chem., 84, 561 (2012).
113) H. Peng, L. Xu, L. Zhang, K. Zhang, Y. Liu, H. Wu, and P. Wu, J. Mater. Chem., 22, 14219 (2012).
114) L. Xu, N. Li, H. Peng, and P. Wu, ChemCatChem, 5, 2462 (2013).
115) H. Peng, L. Xu, H. Wu, K. Zhang, and P. Wu, Chem. Commun., 49, 2709 (2013).