多孔性金属錯体によるナノ空間の科学Nanospace Science of Porous Metal Complexes
名古屋大学大学院工学研究科Graduate School of Engineering, Nagoya University
受理日:2016年2月1日Accepted: February 1, 2016
発行日:2016年4月15日Published: April 15, 2016
多孔性金属錯体は様々な有機配位子と金属イオンによって構築される結晶性の固体で,内部にミクロ孔領域のナノ空間を有する物質群である。この物質群の特徴は多様な構成分子によって構造的・化学的に異なったナノ空間を設計し,合成できることである。約20年前に多孔性金属錯体が注目を浴びて以来,多くの多孔性金属錯体が合成され,今までにない高い細孔容量を有する物質や,超高選択的な気体吸着を示す物質が開発されてきた。また,有機配位子と配位結合という,構造的自由度の高い素子と結合との組み合わせによって,結晶でありながら空間構造を柔軟に変化させるゲート型吸着という新しい吸着現象も見いだされている。最近では分子認識機能と動的機能を有機的に統合し,ヘモグロビンのような生体分子が行っている選択的分子認識と分子捕捉・放出機構を結晶で実現するに至っている。また,多孔性金属錯体のナノ空間を利用した物性物理への応用展開も進んでおり,多孔性金属錯体のナノ空間は材料的な興味だけでなく,新しい科学の展開の場としても大きく注目されている。本解説記事では多孔性金属錯体の発展の歴史から,最新の研究成果までを筆者らの研究を絡めて紹介する。
Porous metal complexes (which is also called porous coordination polymer (PCP) or metal organic framework (MOF)) with the nanospace in the microporous region are crystalline solids with backbones constructed from metal ions and organic ligands as connectors and linkers, respectively. The most striking character of these porous materials is that we can design and create nanospaces having different chemical and structural features with choosing various building blocks. In this two decade, this material has attracted considerable attention and a large number of compounds that have exceptionally high void volume and unusually selective gas adsorption functions have been reported. In addition, it is found that some flexible compounds showed gate opening type abrupt gas uptake at certain pressure, which has not been realized in the other porous compounds. Recent researches proved that two functions, molecular recognition and structural transformation, can be integrated in a framework, realizing highly sophisticated functions of bio-molecules such as hemeprotein. In this article, we show history and development of porous metal complexes, and representative functions of porous metal complexes focusing on the authors' materials.
キーワード:多孔性金属錯体;ナノ空間;ガス吸着;ガス分離;物性転換
Key words: Porous coordination polymer; Metal organic framework; Nanospace; Gas adsorption; Conversion of physical property
© 2016 一般社団法人日本ゼオライト学会© 2016 Japan Association of Zeolite
19世紀から始まった有機合成化学は,炭素と水素を中心に,ヘテロ元素を組み合わせ,膨大な数の分子を生み出してきた。分子とは結合電子を骨,元素を節とした「骨格」である。その後20世紀に隆盛した超分子化学は分子を構成単位として,高次の「骨格」を構築することを可能にし,1分子では不可能な,集合構造に基づく機能を人工的に実現可能であることを示した。固体化学の分野では,X線結晶構造解析をはじめとした観察技術の急速な発展によって,物質の「骨格」構造を可視化し,「骨格」と「物性」の相関を解明することが可能となった。このように考えると,合成化学者をはじめとした多くの化学者にとって,前世期の物質化学のパラダイムは元素あるいは分子で作られた「骨格」の化学であったといえるかもしれない。
一方,ゼオライトや活性炭などの固体内部のナノサイズの空間が主要な機能を示す物質にとって,注目すべきは物質の「骨格」以上に,その周りの存在する「空間」であろう。本解説記事の読者の方々には釈迦に説法となり恐縮であるが,あえて強調するために述べさせていただくと,ナノサイズで規定された「空間」(ナノ空間)の世界では,「空間」に取り込まれたゲスト分子の電子状態や分子構造,分子集合構造は,「空間」の大きさや構造に依存した摂動を非常に強く受けることになる。したがって,原理的には「空間」の設計次第で,ゲスト分子の状態をさまざまにコントロールすることが可能となる。例えば,バルクでは見られない分子クラスターや分子ワイヤー等の特異な分子凝集状態を作り出すことが可能であるし,また,疑似的な圧力効果によって,平衡状態や最安定状態から逸脱した不安定状態を作り出し,特異な物性を引き出すことも可能である1)。さらに,それらの凝集およびストレス状態から,エネルギー変換や触媒反応を進行させ,有用な分子変換の場として利用できることは広く知られている。本解説記事で焦点を当てる多孔性金属錯体はその「ナノ空間」を有する物質群の1つである。この物質は1990年ころから盛んに合成され始めたもので,さまざまな有機配位子と金属イオンを用いて骨格を形成し,多様な空間を創造することができることから(図1),空間構造設計の自由度が高く,様々な独自の機能を創出できるものとして期待され,近年盛んに研究されている。本解説記事では,ゼオライト,活性炭に続く第3の機能性ナノ空間物質として期待される多孔性金属錯体の作る,新しい「ナノ空間の科学」の一部を紹介したい。
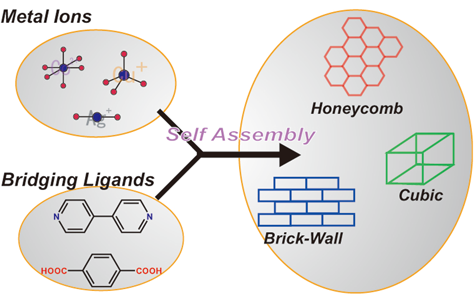
本解説記事で多孔性金属錯体と呼ぶ化合物は,新しい物質であるため,まだ呼称が統一されておらず,日本語では多孔性金属錯体の他に,多孔性配位高分子,多孔性ネットワーク錯体,金属有機骨格と呼ばれたり,英文でもPorous Coordination Polymer(PCP)やMetal Organic Framework(MOF)と呼ばれたりしている。様々な立場から,呼び方や定義が異なっているが,ここでは細かな定義をすることなく,単に結晶性の金属錯体のうち,固体内部にナノ~サブナノメートルオーダーの空間を有する物質を多孔性金属錯体として紹介する。
多孔性金属錯体と呼べる物質は1959年に日本において初めて報告されているが2),急激に注目を集めるようになったのは,1990年以降であり,特に錯体化学の分野において,指数関数的に報告が増え,最近では,錯体化学の分野だけでなく,有機化学,物理化学,計算科学,プロセス化学,物性物理の分野からの研究報告も増加している。上述したように多孔性金属錯体は金属カチオンと,それを連結する有機配位子によって構成される結晶性の固体であり,非常に多様なナノ空間構造を有する物質が合成されている3,4)。そのナノ空間の大きさに関しては,一部のものは2 nmを超えるメソ孔領域の大きさの細孔を有するものも存在するが,ほとんどがマイクロ孔(<2 nm)に分類される細孔を有している。
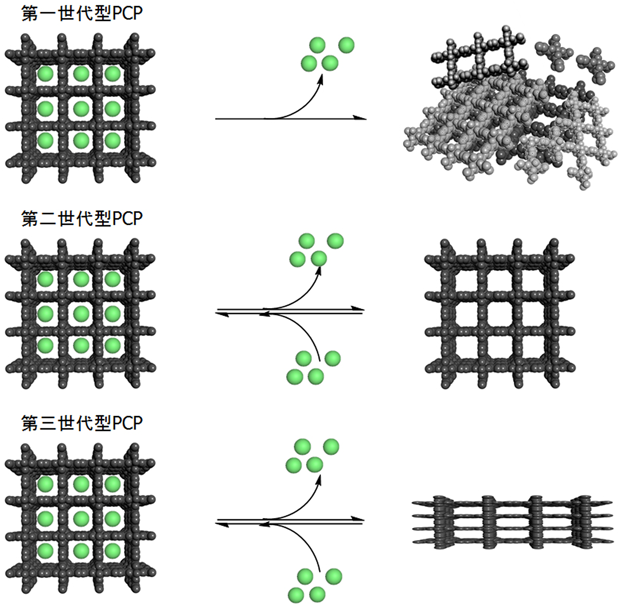
一般に,この多孔性金属錯体は溶媒などのゲスト分子が細孔内に充填された状態で単離される。細孔を利用するためには,減圧,加熱などの操作を行い,このゲスト分子を除かなければならない。多孔性金属錯体のうち,合成時のゲスト分子を除去する操作により崩壊する物質を第一世代型,安定な空間構造を持ち,ゲスト分子を除去してもそのナノ空間を利用できる物質を第二世代型,ゲスト分子を除去したり,ゲスト分子を交換する際,ダイナミックなナノ空間構造の変化を伴うものを第三世代型として分類されている(図2)5)。多孔性金属錯体の研究は骨格構造の多様性に注目した第一世代型を中心とする構造化学的な研究に起こり,その後次第に“第二世代型”つまり安定な細孔構造を利用した分子吸蔵機能の向上を目指した研究へとその中心を移していった。現在では学問的に活発に研究が行われるだけでなく,いくつかの物質は燃料タンクの充填材料等,産業利用に向けた試験が最終段階に入っている。その後2000年代になり第三世代型の多孔性高分子が報告された6)。第三世代型の動的構造変化の起源は,多孔性金属錯体の骨格中の有機配位子の構造的自由度や,配位結合の柔軟性と多様性によるものである。したがって,構造の動的性質はまさに金属錯体材料ならではの性質といえる。現在では,1)安定かつ高細孔容量の構造体の開発,2)高選択的吸着等の高機能物質の開発,3)動的構造に由来する機能の発現というテーマが盛んに研究されている。いずれも従来の無機物や有機物単独では難しい,多孔性金属錯体ならではの機能発現を目指したものであり,“新機能物質系”として研究が展開されている。
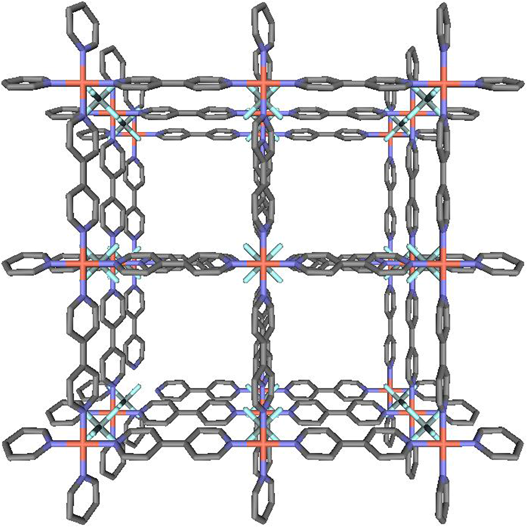
“第二世代型”構造体の構築は90年代中ごろからの多孔性金属錯体の中心的なテーマの1つであった。先に述べたように,多孔性金属錯体は通常,合成溶媒がゲスト分子としてナノ空間に充填された状態で単離される。ゲスト分子と骨格の間に強い化学結合がないことを理由に,これを「ゲスト」と呼びつつも,ゲスト分子は当然骨格とファンデルワールス相互作用や,水素結合によって骨格と相互作用しており,構造体の一部となって構造を安定化している。したがって,このゲスト分子を取り除くと構造は不安定化し,その不安定化に耐えられずに骨格が崩壊してしまうものがほとんどであった。実際,一般に共有結合より弱い配位結合でつくられた構造体が,構造壁の一部となっているゲスト分子を取り除いても安定に構造を維持できるものが存在するとは信じられていなかった。そのため多孔性金属錯体は発見から40年間近くもの間放置されていたのではないかと思われる。さて,そのような先入観が常識となっていた中,北川らは1997年にチャンネル内にゲスト分子が存在しない状態でも安定にその構造が維持され,かつ可逆的にメタン,窒素,酸素ガスを吸脱着する多孔性金属錯体を報告した7)。この物質は4,4'-ビピリジン(bpy)と2価の金属イオン(M=コバルト,ニッケル,亜鉛)と対イオンのナイトレート(NO3−)で構築される物質で,方向の異なる2種類の一次元鎖が柱(ピラー)配位子となる4,4'-ビピリジンで架橋された2次元シートを形成している。この2次元シート構造がかみ合うように積層し,いわゆるTongue–And–Groove構造をとり,隙間にナノ空間がある構造となっていた。この多孔性金属錯体は25°C,30気圧でメタンを約2.3 mmol吸着し,構造と吸着機能の両面から初めて第二世代型の多孔性金属錯体として認められる物質となった。当時は分子(メタン,水素など)を多孔性物質へ貯蔵する技術が,天然ガス利用や水素エネルギーシステムの実現に向けたキーテクノロジーとして大きな注目を集め始めた時であったため,この報告は一躍注目あび,これをきっかけに,以後,安定で高い空隙と表面積を有する多孔性金属錯体の構築とガス吸着能に関する研究が世界中で行われることとなった。2000年には銅(Ⅱ)イオンと4,4'-ビピリジンで構成される2次元シートをヘキサフルオロシリケートアニオン(SiF62−)が連結したジャングルジム型の構造を持つ三次元の多孔性金属錯体を報告した(図3)。この多孔性金属錯体は,比表面積が1500 m2/gを示し,メタン吸蔵量は25°C,35気圧で6.5 mmol/gであった8)。この多孔性金属錯体の出現によって,メタン吸蔵材料の実用化が急速に現実味を帯びて来る事となった。また,メタン吸蔵に向けた多孔性金属錯体の設計指針として,1)構造の次元性を上げ,三次元構造を配位結合で頑丈に組み上げること,2)細孔の壁を薄くして無駄の無い細孔空間を実現すること,3)メタンが充填するのにちょうど良い細孔サイズ(約12×12 Å2)を実現することなどの基本的設計指針を与えることになった。2001年には,三次元のジャングルジムタイプの骨格を持つ集積型金属錯体([Cu2(O2C–R–CO2)2(ted)]n)が関らによって合成された9)。この錯体は,ジカルボン酸の長さによって5~14 Åの細孔を有する一連の多孔性骨格を構築することができる。配位子がビフェニルジカルボン酸(細孔サイズ14 Å)を用いた錯体では,その比表面積が3300 m2/gにも達する10)。これは理想的な活性炭の理論最高値2630 m2/gを遥かに上回る驚異的な値である。このような驚くべき表面積の大きさは,細孔を形成する壁がベンゼン環一枚分しかなく構成原子のほとんど全てが表面に出ているという多孔性金属錯体特有の細孔構造によるものである。
Yaghiらのグループは亜鉛4核クラスター(Zn4O)を種々のカルボン酸で連結した第二世代型の多孔性金属錯体を合成し,特筆すべき研究結果を報告している。1999年に最も注目を浴びた化合物は,Zn4Oのクラスターを6つのテレフタル酸で架橋したMOF-5と呼ばれる化合物である11)。単位胞は立方体で8つの格子点のそれぞれに亜鉛4核クラスターが存在しており,立方体の辺にあたる部分に存在するテレフタル酸によって亜鉛クラスターが連結され,無限構造となっている。MOF-5の細孔表面積は2900 m2/gに達し,77 Kにおける窒素の最大吸着量は660 ml (STP)/gとかなり大きな値を示すものであった。彼らはこの研究をきっかけに,次々と連結部位のカルボン酸配位子を変えた化合物を合成し,空隙を大きくしていった。2002年にはトリフェニレンジカルボン酸でZn4Oを架橋した化合物(IRMOF-16)を合成した12)。この化合物はMOF-5よりも立方体の辺の長さがベンゼン2つ分長くなった化合物で驚異的に大きな空隙を有するものであった。その固体の密度は0.21 g/cm3であり,水より断然軽く,無機固体で最も密度の小さいリチウムの密度(0.56 g/cm3)よりもさらに軽く,まさに「すかすか」の固体といえるものであった。このグループは配位子の組み合わせを変えることで,高表面積の記録を自らの化合物で次々と塗り替えていき,2004年に報告した化合物(MOF-177)13)は細孔表面積が4500 m2/gであり,77 Kにおける窒素吸着量は1080 ml (STP)/g, 2008年に報告したMOF-205の1410 ml (STP)/g, 2010年に報告したMOF-210は2330 ml (STP)/gとなっている14)。
このZn4Oクラスターをはじめとする亜鉛酸素クラスターをコアとしたMOFシリーズの多孔性金属錯体は驚異的な空隙体積を与える一方で,構造としては脆弱であり,湿気や酸には非常に弱く,壊れやすい。大きな空隙を有しながら,酸や空気にも安定であるという事に注目するとMILと呼ばれるシリーズ化合物がある。Féreyらによって2005年に報告された多孔性金属錯体(MIL-101)はクロミウムと酸素のクラスターに対し6方向にテレフタル酸が架橋し,無限構造を形成している。その細孔径の大きさは2~3 nmにもなり,細孔の大きさは,マイクロ孔領域(<2 nm)を超えてメソ孔(2 nm<n<50 nm)に分類される大きさでありながら,空気中で非常に安定で酸にも強い15)。これは,合成溶媒にフッ酸水溶液や硫酸水溶液を用いて合成することによって,酸や水に耐性のある多孔性金属錯体を合成するというアイディアによるものであると考えられる。このMILシリーズの多孔性金属錯体はその高い安定性から触媒やヒートポンプ等への応用利用が盛んに研究されている。
このように第二世代型の多孔性金属錯体の研究は,1997年にはじまって,高空隙率,高内部表面積を競う研究はいまだ続いており,まだ天井を見せていない。高表面積を有する物質は大量に分子を吸着できる吸蔵材料としての応用展開に魅力があり,そこに研究のモチベーションがある。多孔性金属錯体を吸着材料として,水素やメタンなどの燃料ガスを大量に吸蔵し,運搬する研究も進んでおり,これらが実用化されれば大量エネルギー消費時代の環境問題を解決する助けになるものと大きな期待が寄せられている。
多孔性金属錯体が他の物質と比較して,特徴的な点の1つは,有機分子を骨格に用いており,この有機分子の性質や機能をナノ空間に直接反映させられることである。例えば,有機分子にヒドロキシル基を導入し,ナノ空間の親水性を制御したり,有機分子に不斉点を導入することで,ナノ空間をキラルな場にしたりすることが,容易に可能である16,17)。また,もう1つ重要な点は,多孔性金属錯体は結晶であり,導入した官能基は規則的で,均一に細孔表面に存在しているという点である。官能基と効果的に相互作用できるゲスト分子は,官能基の配列と同調した規則的な配列で吸着されることが期待できる。この際,ナノ空間の立体的な構造が特定のゲスト分子とうまく合致すれば,ゲスト分子の吸着状態は均一で,吸着エネルギーの分散は小さくなる。すなわち,特定の分子に対して特異的に,強い束縛状態を実現できると期待される。このことを利用して,様々なゲスト分子に対して,それぞれ特異的に吸着する多孔性金属錯体が開発されてきており,例えば工業的に重要なキシレン類の異性体の一つを特異的に吸着して分離するような多孔性金属錯体も開発されている18,19)。本節では,このような特異的な選択性を示す多孔性金属錯体の1例として,著者らの開発したアセチレン高選択吸着を示す多孔性金属錯体の研究例を紹介する。
2価の銅イオンと2,3-ピラジンジカルボキシレートおよびピラジンで合成されるCPL-1と呼ばれる多孔性金属錯体は4×6 Å2の非常に小さな1次元細孔を有している。CPL-1の合成時のナノ空間中には,水分子が充填されており,この水分子は細孔表面に露出している配位子由来の塩基性の酸素原子と水素結合し,細孔内で水の一次元ワイヤーを形成している。この酸素原子と相互作用できるゲスト分子は特徴ある吸着挙動を示すと考え,ゲスト分子の検討を行い,アセチレンを候補とした。アセチレン分子は,両末端に比較的酸性な水素原子を有しており,細孔壁の塩基性の酸素原子と酸–塩基の相互作用が予想される。また,アセチレンは非常に反応活性な分子で,たった2気圧の加圧で分解爆発の危険性のある分子として知られている。もしこの非常に反応活性なアセチレン分子を安定に細孔中へ吸着,濃縮できる事を示せられれば,多孔性金属錯体ならではの高機能表面を利用した吸着機能となると考え,アセチレンの吸着実験を行った20)。
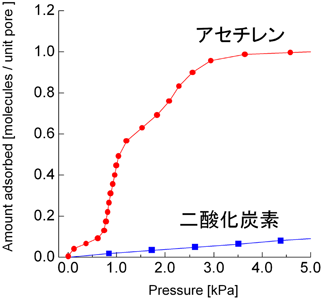
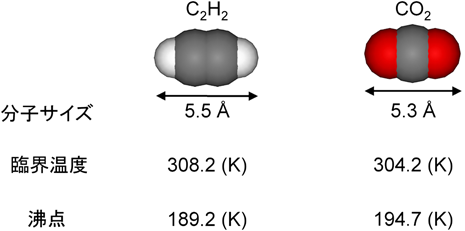
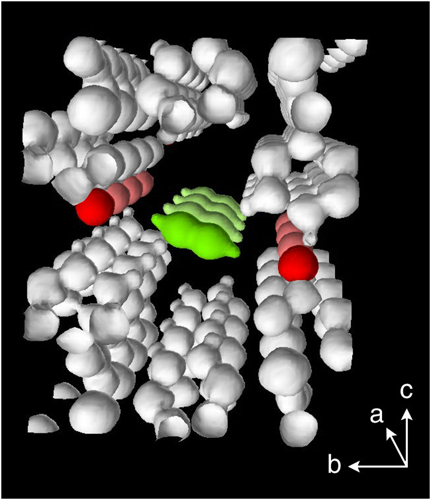
図4は270 KにおけるCPL-1へのアセチレンと二酸化炭素の吸着等温線である。二酸化炭素はアセチレンとサイズおよび沸点などの物理化学的性質が酷似しているため(図5),通常の吸着剤では同様の等温線のプロファイルを示す。しかしながら,CPL-1の吸着では,アセチレンは二酸化炭素と比べて,かなり低圧で一気に吸着が起こるため,アセチレンと二酸化炭素の吸着量の差は最大で26倍量にもなることがわかった。さらに等量吸着熱もアセチレンの方が10 kJ/molも大きく,吸着によるアセチレンの安定化が顕著に示された。そこで,このアセチレンの吸着現象を構造的に解明する事を目的に,粉末X線回折測定とMEM(Maximum Entropy Method)/Rietveld法21)と呼ばれる解析手法を用いて,吸着前後における細孔構造を明らかにした。その結果,興味深いことに,棒状のアセチレン分子はその向きを細孔壁の酸素原子の方へ向け,あたかも酸素原子に抱えられているように取り込まれていた(図6)。また細孔内部のアセチレンは爆発限界の約200倍もの密度に濃縮されている驚異的な状態であることがわかった。これは,アセチレンが細孔中で壁の酸素原子によって強く束縛され,決まった配向でしか吸着されないため爆発的な反応が起こらないものと考えている。
これまでは安定な骨格とその内部のナノ空間を利用する事を基盤にした,第二世代型の多孔性金属錯体を紹介してきた。ここでは動的なナノ空間を有する第三世代型の多孔性金属錯体について紹介したい。多孔性金属錯体は,配位結合,水素結合,π–π相互作用,ファンデルワールス力などで作られており,ゼオライトや炭素材料を支える,Si-O(または/Al-O)結合,C-C共有結合などと比較してそれほど強くない。加えて,有機配位子の柔軟性,たとえばメチレン鎖のゆらぎやベンゼン環の回転などの自由度も存在する。つまり,多孔性金属錯体のもともと「柔軟性」に富んだ物質であるといえるが,第二世代型の剛直な多孔性金属錯体を設計する上で,この「柔軟性」は欠点であり,「柔軟性」を生かした物質設計は,当初考えられることはなかった。しかしながら,ゲスト分子の吸脱着によって可逆的にその骨格とナノ空間構造を変化する多孔性金属錯体が発見され,一躍「第三世代型」が注目を浴びるようになった。本項目では第三世代型の多孔性金属錯体を設計する上で重要な3つの要素を示し,それぞれ例を挙げて紹介する。
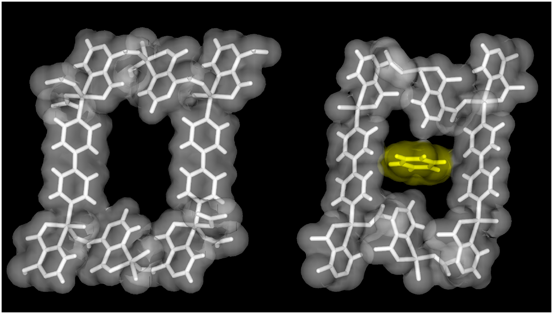
先にCPL-1という多孔性金属錯体を紹介したが,この化合物の構造中の柱配位子をピラジンから,倍の長さの4,4'-ビピリジンに変えると空間サイズが6×8 Å2程度の一次元細孔を有するCPL-2という化合物が合成される。このナノ空間のサイズはベンゼンがアクセスできる程度の大きさであるが,特に細孔表面にベンゼンと強く相互作用するような官能基があるわけではない。しかし,吸着実験を行うと1細孔あたりきっちり1分子のベンゼンを吸着する興味深い結果が得られた。この吸着構造の結晶構造解析を行うと,確かに1細孔あたり1分子がきっちり入っており(図7),しかも興味深いことに吸着の際にナノ空間は約5%収縮しており,ベンゼンに対して,相互作用の大きさが大きくなるように空間構造を収縮させ,誘導適合型の変化を示す事が分かった。これはゲストを取り込んで膨張する多くの第三世代型の化合物と逆の性質であり,驚くべき結果であった。このナノ空間の収縮の起源が何かという事であるが,実は配位結合の開裂と生成に基づいている。2価の銅イオンは4配位平面構造,5配位四角錐構造,6配位八面体構造をとりやすいが,アキシャル位はヤーンテラー効果によって結合が弱く,配位環境の違いによって容易に配位数は変化する。CPL-2のベンゼンの取り込みの際の空間構造変化はまさに5配位四角錐構造から,4配位平面構造への配位構造の変化を伴っており,配位結合の柔軟性によって動的空間を実現した好例といえる22)。
アミド基は水素結合を形成する代表的な官能基である。このアミド基を有する配位子N-(3-ピリジル)イソニコチンアミドと2価のコバルトイオンで合成した多孔性金属錯体は水素結合性のTHFやアセトンなどの吸脱着に伴って,結晶の色がピンクから紫へと変色する。この多孔性金属錯体はアミド基が骨格の2次元シートの面内でゲスト分子と水素結合を形成する。一方,ゲスト分子が脱着するとアミド基が面と垂直方向に回転して2次元シート間で水素結合ネットを形成し,水素結合と配位子の回転運動が連動する機構が働いている。したがって,ゲストの吸脱着の際,水素結合の生成と開裂を起源とする,空間構造変化が可能であることを示している23)。
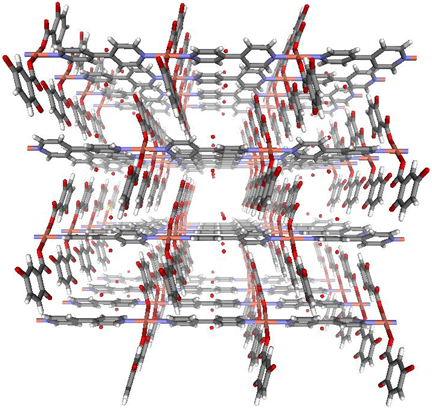
2,5-ジヒドロキシ安息香酸(dhba)と4,4'-ビピリジンを用いて得られる銅錯体は,2次元シートがdhba部位でπ–πスタッキングをした積層構造を有している(図8)24)。骨格自体は剛直な2次元シートであるが,シート間に直接の結合がないため全体の3次元構造は非常に柔軟性に富んでいる。実際,この多孔性金属錯体は合成時の細孔に取り込まれている水分子を取り除くと細孔が収縮し細孔が無くなってしまう。しかし,この多孔性金属錯体は超臨界流体の吸着によっても劇的な構造変化が起きる。298 Kにおけるメタン吸着実験では,8気圧までまったく吸着を示さないのに対して,8気圧を境にまるで閉じていたゲートが開いたかの様に急激な吸着を示す(図9)。また,吸脱着には明確なヒステリシスが存在し,この挙動は細孔の無い構造から多孔性構造への結晶構造変化であることが明らかにされている。現在ではこのような吸着現象をゲート型吸着25)と呼び,数多く報告されている。ゲート型吸着では小さな圧力操作で吸着分子を大量に吸脱着できることから,気体貯蔵材料への応用が期待されている。
生体内血液中にあるヘモグロビンタンパク質は非常に効率よく酸素分子を運搬していることが知られている。ヘモグロビンはその構造中に4つの酸素と相互作用可能な鉄イオンサイトを有しているが,4つのサイトが独立に酸素を取り込むのではなく,1つの部位が酸素を取り込むと,残りの3つの部位が酸素を取り込みやすくなるように,タンパク質全体の形が協同的に変化する。すなわち,表面官能基の分子認識機能と,空間構造の動的機能が統合した協同的機能によって,効率よく酸素を捕捉したり放出したりしている。動的機能と表面機能を統合した協同的機能を有する多孔性金属錯体を合成できれば,生体が行っている効率的な分子の吸脱着機能を実現できると期待できる。このような協同的機能を一酸化炭素分子(CO)に対して示す多孔性金属錯体の例を1つ紹介する。
ここで挙げる多孔性金属錯体は2価の銅イオンとイソフタル酸誘導体で合成される2次元シート骨格を有する多孔性金属錯体である26)。この多孔性金属錯体はカゴメ格子構造を有しており,六角形の大きな蛇腹型1次元チャンネル(L細孔)と三角形の小さな蛇腹型1次元チャンネル(S細孔)が積層した2次元シートを貫くよう存在している(図10)。まず結論を述べてしまうと,この2つのチャンネルのうちL細孔は特別の機能は持たないが,S細孔が協同的機能を示す。
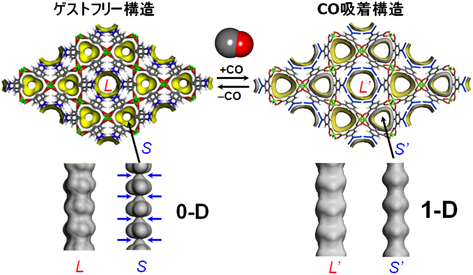
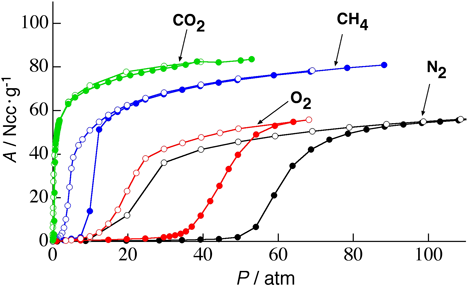
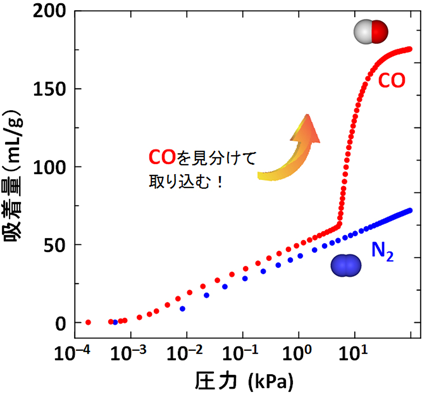
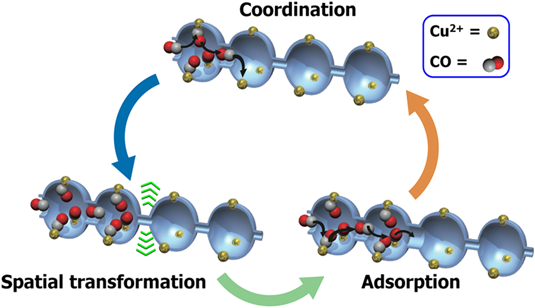
まずゲストを取り除いた構造では,S細孔は蛇腹構造の狭くなった部分が2原子分子のCOや窒素分子(N2)でも通過できない大きさとなっている。また,2価の銅イオンサイトが細孔表面にあるが,このサイトは上下のシート内に存在する酸素原子と結合しており,銅の配位構造は大きく歪んだ5方四角錐構造となっている。一見するとこのS細孔にはCOもN2も何も吸着されないように予想されたが,実際に吸着実験を行うと(図11)のようなCOに対してだけ選択的にゲート型吸着を示すことが分かった。詳細は割愛するが,その原理は次のように理解されている。CO分子は,結晶表面の銅イオンサイトに認識され,骨格の酸素–銅結合を開裂し,それが駆動力となり,蛇腹の狭い細孔を押し広げて細孔Sの1つ奥の細孔に取り込まれる。この奥に入ったCOは同様に表面に存在するCuサイトに再度認識され,骨格の酸素-銅結合を開裂し,蛇腹を開いてさらに1つ奥の細孔へと押し込まれる。このサイクルを繰り返すことによって,等温線にあるような急激なCOの取り込み(自己加速的な吸着プロセス(図12))が実現されている。一方,N2はCuによって認識されないので,全く蛇腹が開かず取り込まれない。すなわち,この系は分子を認識する表面機能と,動的な構造変化機能が統合した,協同的機能を有しており,まさに生体分子が行っているような,高度な分子認識と運搬機能を結晶性でも実現可能であることを示すものである。補足するとこの物質はCOとN2の混合ガスから実際にCOを分離することが可能で,応用的発展性が期待されることから科学技術振興機構のACCELプロジェクトの1つテーマとして研究が進んでいる。
ここまで紹介した研究は多孔性金属錯体を気体の分離や貯蔵を見据えた材料研究であったが,多孔性金属錯体のナノ空間を使った興味深い基礎的な物性研究も行われている。多孔性金属錯体のナノ空間はかなり自由に構造を設計し構築できるので,内部に取り込まれる分子のクラスターのサイズや構造,クラスター内やクラスター間の相互作用の大きさ,といった構造的・物理的・化学的なパラメータを思い通りに制御できる。従来,物性物理の分野では,バルク物質を極低温や超高圧にすることで,このようなパラメータを制御し,新たな物性探索の研究が広く行われてきた。しかし,目的のクラスターを意図的に作り出すことは極めて難しく,多くの場合偶然の発見に依存している。それに対し,多孔性金属錯体のナノ空間は,意図的に空間を設計することができるため,ナノ空間内で目的の分子クラスターを作り出すことができる。また,バルクでは分子クラスターを孤立させることも難しいが,ナノ空間を利用すれば,理論的な予想しかできなかった分子クラスターを現実世界で実現できる。本項目では,多孔性金属錯体のナノ空間に分子を規則配列させた新しい分子クラスターが示す新奇な物性に関する研究を紹介する。
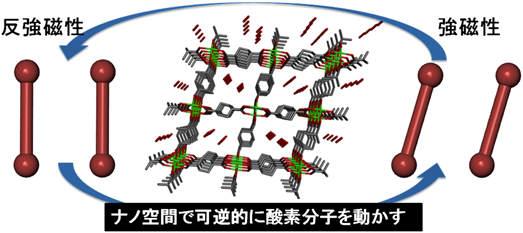
磁性分子を空間的に自由に配置し,その磁気的な性質を観測することは磁性分野での大きな研究テーマの1つである。例えば,三角形の頂点に磁性分子や原子を配置し,それぞれのスピンが反強磁性結合をもつ場合,スピン配置は乱れて規則状態が実現できなくなる。つまり空間的配置は三角形と美しくてもスピン配置は乱れてしまう。このような磁性分子を特定の位置に配置した磁性分子クラスターの磁気的性質と相転移現象は,大きな注目を集める研究対象である。
まず,先に述べた単純な系としてCPL-1に磁性分子である酸素分子を吸着させた研究を紹介する。細孔サイズは4×6 Å2であるため,この中に酸素は2分子取り込まれ,分子間距離が3.2 Åの孤立した酸素2量体を形成する27)。分子が平行に並んだH型をとり磁気的には反強磁性相互作用が極めて強い状態が観測された。このような二量体において酸素分子間の配向を制御できれば反強磁性から強磁性へと磁気転換し,まるで磁気メモリのように駆動することが理論的に知られているが実験的に確認することは不可能であった28)。著者らは,CPL-1のような固い骨格をもつ多孔性金属錯体ではなく,柔軟な多孔性金属錯体であればそのような,酸素分子2量体の配向を変化させ,分子間相互作用が反強磁性から強磁性まで磁気変換することができると考え,実際に銅イオンとシクロヘキサンジカルボン酸を骨格とする多孔性金属錯体のナノ空間内に酸素分子2量体を孤立させ,実験的にそのような酸素の配向変化と磁性変化を初めて証明した(図13)29)。この実験結果は,酸素の物性研究の場に大きな影響を与え,特に分子配向に着目した強磁性物理の分野の研究が大きく進展している。
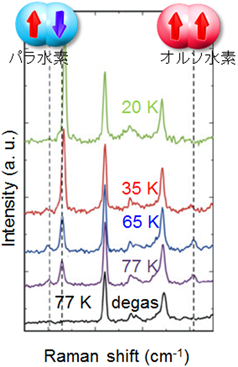
水素分子には核スピンの向きの違いによってオルソ水素とパラ水素の2種類の異性体が存在し,室温では統計学充率によって,オルソ・パラの存在比は3 : 1となっている。パラ水素の方がエネルギー的に安定であるため,低温における熱平衡状態ではパラ水素の方が優先的に存在するが,実際にはこの転換が非常に遅い(転換時間は4.3年)。磁場や電場中に置くと転換が加速されることが知られているがそのメカニズムが十分に明らかにされているわけではない30)。多孔性金属錯体のナノ空間は非常に小さく,細孔表面に金属イオンを露出させると,空間内には大きな電場こう配ができる。堀らはこのことに着目し,ホフマン型の多孔性金属錯体に水素を吸着させ,300秒という通常では考えられないほどの高速でオルソ水素からパラ水素へと転換する事をラマン測定によって観測した(図14)31)。詳細な構造解析の結果,吸着過程で水素分子が電場こう配が急峻な位置へ移動し,オルソ–パラ転換が起こっていることが分かっている。ところで,この水素のオルソ–パラ変換は物性物理においても興味が持たれるものであるが,実は応用的にも重要な側面を持っている。水素を液化して運搬する際,転換時間が非常に遅いため,液化した水素のオルソ・パラ比もはじめは3 : 1となっている。そして,徐々にパラ水素へと転換されていくのであるが,その際発熱し,せっかく液化した水素が蒸発してしまう。これは液化水素のボイル・オフ問題と呼ばれており,これを解決するには気体状態ですべてパラ水素化してしまうオルソ-パラ転換触媒が求められている。ここで紹介した,多孔性金属錯体はこのような応用利用にも期待できるものであり,さらなる研究が行われている。
非常に長い歴史があり,実用物質として使われている,ゼオライトや炭素材料などの多孔性物質と比較して,多孔性金属錯体は遥かに新参者である。そのため,まだまだ未開拓な領域が数多く存在しており,これからも確実に多くの新物質が合成され,新機能や新現象が発見されていくと考えられる。しかしながらその一方で,学術的な興味の対象として進んできた多孔性金属錯体の研究が,実際に我々の生活に役に立つ材料となり得るかどうかというと,まだ確固たる結論が出ているわけではない。今後,他の材料にはない高機能性を生かした,実用材料が生み出されて初めて,本来の意味でゼオライトや活性炭に次ぐ第3のグループの多孔性物質となりうるものである。基礎学術研究がかなり進んできた今,実用材料としての展開が大いに期待されている。本解説を最後までお読みいただいたことに深謝するとともに,多孔性金属錯体の研究開発分野への忌憚のないアドバイスを頂戴できればありがたく思っている。
1) K. Urita, Y. Shiga, T. Fujimori, T. Iiyama, Y. Hattori, H. Kanoh, T. Ohba, H. Tanaka, M. Yudasaka, S. Iijima, I. Moriguchi, F. Okino, M. Endo, K. Kaneko, J. Am. Chem. Soc., 133, 10344–10347 (2011).
2) Y. Kinoshita, I. Matsubara, T. Higuchi, Y. Saito, Bull. Chem. Soc. Jpn., 32, 1221 (1959).
3) S. Kitagawa, R. Kitaura, S.-i. Noro, Angew. Chem., Int. Ed., 43, 2334–2375 (2004).
4) H.-C. Zhou, J. R. Long, O. M. Yaghi, Chem. Rev., 112, 673 (2012).
5) S. Kitagawa, M. Kondo, Bull. Chem. Soc. Jpn., 71, 1739 (1998).
6) K. Uemura, R. Matsuda, S. Kitagawa, J. Solid State Chem., 178, 2420–2429 (2005).
7) M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka, S. Kitagawa, Angew. Chem., Int. Ed., 36, 1725 (1997).
8) S. Noro, S. Kitagawa, M. Kondo, K. Seki, Angew. Chem. Int. Ed., 39, 2082 (2000).
9) K. Seki, Chem. Commun., 1496 (2001).
10) K. Seki, Langmuir, 18, 2441 (2002).
11) H. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Nature, 402, 276 (1999).
12) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O. M. Yaghi, Science, 295, 469 (2002).
13) H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. B. Go, M. Eddaoudi, A. J. Matzger, M. O’Keeffe, O. M. Yaghi, Nature, 426, 523 (2004).
14) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim, O. M. Yaghi, Science, 239, 424–428 (2010).
15) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé, I. Margiolaki, Science, 309, 2040 (2005).
16) T. K. Maji, K. Uemura, H.-C. Chang, R. Matsuda, S. Kitagawa, Angew. Chem., Int. Ed., 43, 3269–3272 (2004).
17) J. Seo, R. Matsuda, H. Sakamoto, C. Bonneau, S. Kitagawa, J. Am. Chem. Soc., 131, 12792–12800 (2009).
18) J. E. Warren, C. G. Perkins, K. E. Jelfs, P. Boldrin, P. A.Chater, G. J. Miller, T. D. Manning, M. E. Briggs, K. C. Stylianou, J. B. Claridge, M. J. Rosseinsky, Angew. Chem. Int. Ed., 53, 4592–4596 (2014).
19) R. Matsuda, Nature, 509, 434–435 (2014).
20) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe, Y. Mita, Nature, 436, 238 (2005).
21) M. Takata, E. Nishibori, M. Sakata, Z. Kristallogr., 216, 71 (2001).
22) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, S. Horike, M. Takata, J. Am. Chem. Soc., 126, 14063–14070 (2004).
23) K. Uemura, S. Kitagawa, M. Kondo, F. Koichi, R. Kitaura, H. Chang, and T. Mizutani, Chem. Eur. J., 8, 3587 (2002).
24) R. Kitaura, K. Seki, George. Akiyama, S. Kitagawa, Angew. Chem., Int. Ed. Engl., 42, 428 (2003).
25) D. Li, K. Kaneko, Chem. Phys. Lett., 335, 50 (2001).
26) H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata, S. Kitagawa, Science, 343, 167–170 (2014).
27) R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, Ho-Chol Chang, T. C. Ozawa, M. Suzuki, M. Sakata, M. Takata, Science, 298, 2358 (2002).
28) B. Bussery, S. Ya. Umanskii, M. Aubert-Frécon, O. Bouty, J. Chem. Phys., 101, 416 (1994).
29) A. Hori, T. C. Kobayashi, Y. Kubota, A. Matsuo, K. Kindo, J. Kim, M. Takata, H. Sakamoto, R. Matsuda, S. Kitagawa, J. Phys. Soc. Jpn., 82, 084703 (2013).
30) R. Muhida, H. Setiyanto, Md. M. Rahman, W. A. Diño, H. Nakanishi, H. Kasai, K. Fukutani, T. Okano, Thin Solid Films, 509, 223 (2006).
31) T. Kosone, A. Hori, E. Nishibori, Y. Kubota, A. Mishima, M. Ohba, H. Tanaka, K. Kato, J. Kim, J. A. Real, S. Kitagawa, M. Takata, R. Soc. Open Sci., 2, 150006 (2015).
This page was created on 2017-03-06T15:18:12.193+09:00
This page was last modified on
このサイトは(株)国際文献社によって運用されています。