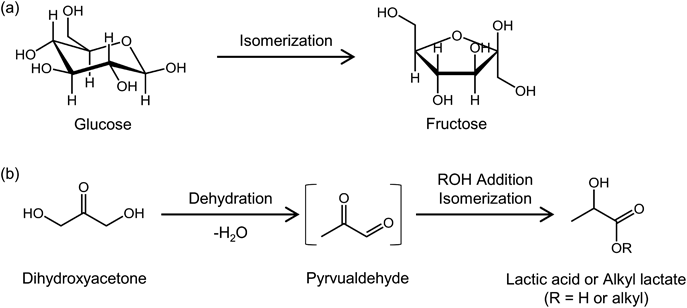
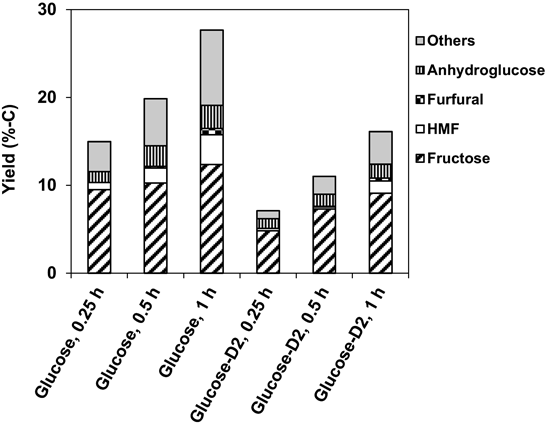
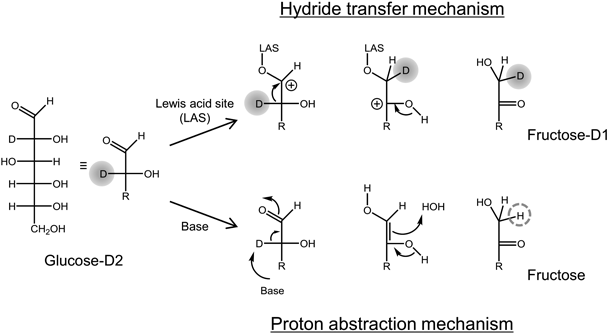
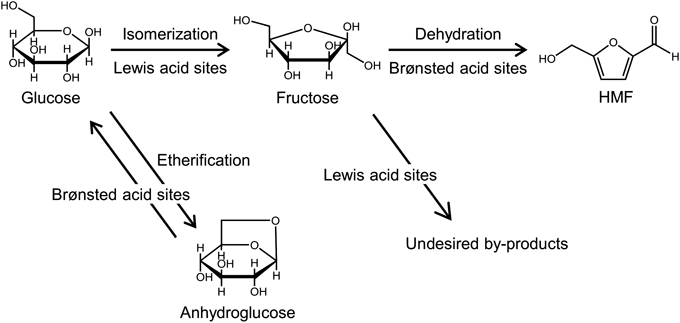
ゼオライトベータを固体触媒に用いた糖類の変換Conversion of Saccharides Using Zeolite Beta as Solid Acid Catalyst
東京工業大学 資源化学研究所Chemical Resources Laboratory, Tokyo Institute of Technology ◇ 〒226–8503 横浜市緑区長津田町4259
受理日:2016年1月7日Accepted: January 7, 2016
発行日:2016年4月15日Published: April 15, 2016