金属イオンと有機架橋配位子を混合し,錯形成・重合を行うことによって,結晶性の金属錯体集積物が合成されている。このような材料群を金属有機構造体(Metal Organic Framework: MOF)あるいは多孔性配位高分子(Porous Coordination Polymer: PCP)と呼んでいる。MOF/PCPは自己組織化型の多孔性材料としても大きな注目を集めており,活性炭やゼオライトをはるかに超える高比表面積を有する。結晶性であるため,そのナノ空間は高い規則性と均一性を担保する。また,有機架橋配位子の分子デザインと,金属との組み合わせによって,類をみないほど多彩な構造を設計できる。さらに,最大の特徴は配位結合や柔軟な部位をもつ架橋配位子に由来する構造の柔軟性「やわらかさ」にあり,活性炭やゼオライトなど既存の「かたい」多孔性材料とは一線を画する。このような特徴を科学し,巧く工学することにより,先行の多孔性材料では成しえなかった新しい応用展開が拓かれつつある。MOF/PCPの研究は世界中で急速に展開されており,優に100を超える総説1)があるので,その構造や機能,用途などを改めて紹介するまでもない。本稿では,より一層の簡便性・量的生産性を図るために筆者らが取り組んできた水系合成法の開発と結晶形態の制御を中心に解説し,そこから膜分離への展開における現状と課題を探ってみたい。
ユニークな特徴をもつMOF/PCPにおいても,その水熱安定性は多くの産業用途に適応するためには避けられない課題である。配位結合性のMOF/PCPはゼオライトに比べて物理的・化学的安定性が低く,水に不安定なものが多い。そのような中で,ゼオライト様トポロジーを有するイミダゾレート構造体(Zeolitic Imidazolate Framework: ZIF)は構造安定性が高いことで知られている2,3)。ZIFはMOF/PCPのサブファミリーの一つであり,亜鉛(あるいはコバルト)イオンとイミダゾール系の架橋配位子から構成される。脱プロトンしたイミダゾール類が金属イオンに配位してゼオライトのO–Si–O結合と同程度の角度をなすため,ゼオライトと類似した結晶構造を形成する。中でもSOD型トポロジーを有するZIF-8は構造安定性が高く,その細孔径がガス吸着・分離に応用しやすいことから報告例が最も多い。その合成は,MOF/PCPと同様,有機配位子と金属イオン間の配位結合・錯形成を基本とするため,有機液相で固体析出させる手法が一般的である。硝酸亜鉛や塩化亜鉛などの亜鉛塩と2-メチルイミダゾール(Hmim)をN,N-ジメチルホルムアミドやメタノールなどの有機溶媒中でソルボサーマル反応させることにより,単結晶状のZIF-8粒子が得られる。しかし,有機溶媒の使用は,その揮発性,可燃性,毒性による安全上の問題や環境への悪影響のみならず,生成物の細孔内にバルキーな有機溶媒分子が残存することにより活性化に手間がかかることが問題となる。
MOF/PCPが先行のゼオライトに対して工業的優位性で肩を並べる影響力をもつためには,機能性材料として精密に構造設計する技術に加えて,環境・コスト面でも有望な製造技術を体系化することが重要である。筆者らは,ZIF-8の架橋配位子であるHmimが比較的水によく溶解する(20°Cにおける水への溶解度780 g/l)ことに着目し,ZIF-8の水系合成法を検討してきた4,5)。
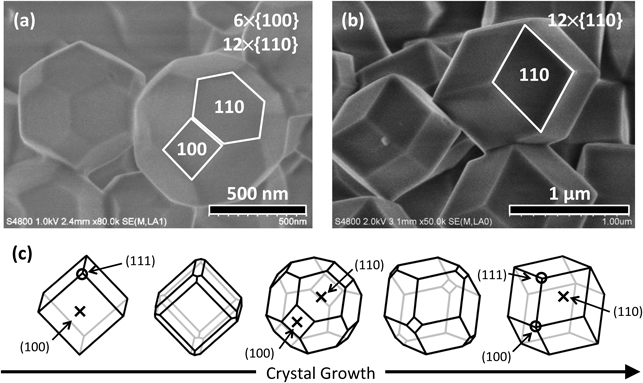
水系合成は至ってシンプルで,常温・常圧下で亜鉛塩とHmimの水溶液を混合する方法である。硝酸亜鉛を用いれば,ものの10秒で混合液は白濁し,20分のうちに亜鉛基準で90%を超える粒子収率が得られる。生成物は切頂菱形12面体(6×{100}面と12×{110}面の計18面体)の晶癖(形態)を特徴とする(Fig. 1a)。水系合成で得られたZIF-8は従来の非水系で調製されるものと比べて同程度あるいはそれ以上の比表面積(BET比表面積〜1700 m2/g)ならびにミクロ孔容積(〜0.65 cm3/g)を示し,清浄な界面を有していることがわかる。TGA測定からZIF-8は空気中で400〜450°Cまで安定であると見積もられ,熱分解による重量減少率(ZIF-8; Fw=229.6, ZnO; Fw=81.4)から生成物の組成上の純度は100%に近いことを確認することができる。
水系合成法では,亜鉛塩の種類や合成温度,Hmim/Zn比を調整すれば,生成する結晶の粒子径と晶癖(形態)を制御できる。例えば,合成温度を25°Cから70°Cに上げると,晶癖(形態)は切頂菱形12面体から菱形12面体に,粒子径は0.73 µmから1.5 µmに変化する(Fig. 1b)。有機液相でのZIF-8結晶の成長過程はCravillonらによって報告されている6)。ZIF-8の{100}面は最も表面エネルギーが高く,成長速度の速い結晶面である。{100}面の露出は熱力学的に不安定であり,遅い結晶成長速度をもつ{110}面が結果として大きな面として現れる(Fig. 1c)。ZIF-8の最安定相は菱形12面体であるが,25°Cの水系合成では反応速度が速く,反応初期の核生成量が多くなるため,結晶成長を促す原料養分が不足することによって粒子径が小さく,かつ切頂菱形12面体で成長が終結したと考えられる。一方,生成した核の特定の面に吸着する物質(界面活性剤やイオンなど)をキャッピング剤として添加することにより,その結晶面の成長速度を抑制することで晶癖(形態)を制御することもできる。
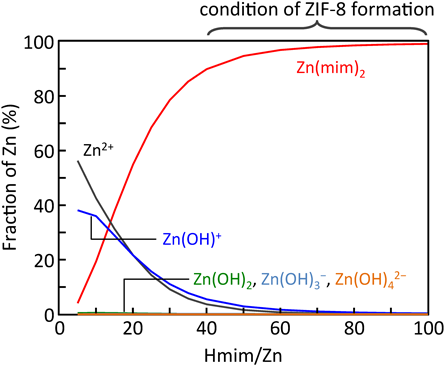
水系合成法では有機溶媒の使用を大きく低減することができる一方,ZIF-8骨格のHmim/Zn化学量論比よりも過剰量のHmimを必要とすることがわかった。SOD型トポロジーのZIF-8の骨格はHmim/Zn=2で構成されるが,水系合成法では濃度依存性はあるがおおむねHmim/Zn≧40の条件がZIF-8の生成に必要になる。このことは錯体の安定度定数である程度説明できる。水系合成では金属–配位子と金属–水分子の配位競合のため,Hmim濃度が低いと亜鉛の水酸化物塩(例えば,Zn(OH)(NO3)(H2O)など)が副生成物(不純物)として析出してしまう。ZIF-8錯体の安定度定数(KZIF8)は未知数であるが,反応系内の亜鉛錯体と配位子の物質収支(eq. 1, 2)と電荷均衡(eq. 3)を連立し,反応溶液のpHをフィッテイングパラメーターとして実測pHに一致するようにKZIF8を求めた。推算したKZIF8を用いることにより,反応系内の化学種の濃度分布を予測した。KZIF8の推算は,eq. 2で示す通りZn(mim)2をZIF-8とし,Zn(OH)i2−i(i=0~4)あるいはZn(mim)2が生成するという簡潔な仮定によるものであるが,化学種の濃度分布,ZIF-8の生成範囲を実によく表現することができている(Fig. 2)。このように,MOF/PCPの生成条件は錯体の安定度定数のデータベースを拡充させることにより,容易に予測することができるだろう。
物質収支
電荷均衡
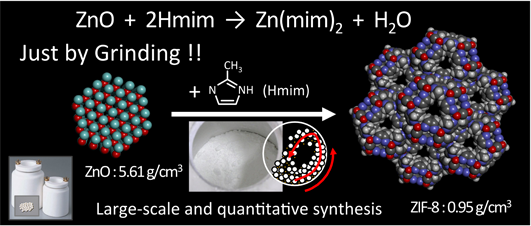
MOF/PCPの生成プロセスは錯形成反応であるため,液相中での合成が一般通念である。筆者らは環境負荷の低減を目的とし,非水系で合成されてきたZIF-8をそれと同品質以上で水系合成できることを実証した。その一方で,水系合成では金属–水分子の配位結合のために,非水系に比べて過剰量のHmimの添加が必要であるという課題が提示された。そこで筆者らは,いっそのこと無溶媒でZIF-8を合成することを目論み,その手法としてメカノケミカル法に注目した。メカノケミカル合成は至ってシンプルで,常温・常圧下で酸化亜鉛とHmimの粉体混合物をボールミルで機械的に混合する方法である7)。XRD,N2吸着,TGA測定から,転動ボールミルによる混合時間とともに非多孔性の酸化亜鉛が多孔性のZIF-8に結晶転換していく様子を確認することができる。転動式のボールミルは構造が簡単かつ安価で大規模な粉砕機へのスケールアップも容易であり,微粉体の大量生産に適している。一方,ボールと原料の重力による混合のため,反応に長時間を要する。遊星式のボールミルを用いれば,反応容器の自転とターンテーブルの公転の組み合わせによる強い遠心力を利用することができ,反応時間を短縮することができる。
本手法では,溶媒が不要であり,酸化亜鉛とHmimを化学量論比Hmim/Zn=2で機械的に混合するのみでZIF-8が生成する。また,副生成物は水のみであり,固液分離を必要としないため生成物のロスが少なく,大量生産が可能であるため,工業化にも適している。亜鉛源として溶媒に溶けにくい酸化亜鉛を出発原料として利用できることも本手法の大きな特徴であり,溶媒に溶けにくい架橋配位子を用いることができる点で水系合成法における課題も克服できる。メカノケミカル処理は,結晶性物質に衝撃・圧縮・摩砕などの機械的エネルギーを作用させ,新生界面の形成とともに継ぎ手を失った表面原子・分子数を増大させることにより,物質を活性化する手法であり,非晶質化,相転移,他の物質との反応性,吸着性の増大などの現象が現れる8,9)。これまでにも多孔性材料の合成にメカノケミカル法が利用されてきたが,無機系材料のゼオライト合成における前駆体原料の反応性を向上させるための前処理10)あるいはゼオライト結晶をナノ粒子化させるための粉砕手段11)としての利用に限られている。いずれの場合もメカノケミカル処理後に,水熱条件下でのゼオライトの結晶化プロセスが欠かせない。これに対して,ZIF-8合成におけるメカノケミカル処理は,多孔性ナノ空間の直接的な形成経路を担っており,非多孔性の酸化亜鉛(ρZnO=5.61 g/cm3)から多孔性のZIF-8(ρZIF8=0.95 g/cm3)に低密度化(体積膨張)しながら結晶転換される点が非常に興味深い(Fig. 3)。
メカノケミカル法で得られるZIF-8の特徴の一つは,3.で示した単結晶状のZIF-8に見られるような特定の結晶面をもたないところにあり,一見アモルファス状の一次粒子が凝集した二次粒子として得られる(Fig. 4a)。XRD測定から,反応後にも酸化亜鉛が残存していることが推察され,生成物の熱分解による重量減少率から酸化亜鉛のZIF-8への転化率は80%程度と見積もられる。つまり,20%の酸化亜鉛が生成物中に残存していることになるが,未反応の酸化亜鉛を含む生成物の単位質量あたりの表面積(BET比表面積~1500 m2/g),細孔容積(~0.60 cm3/g)は従来の非水系で調製されるものと比べても遜色なく,細孔特性という観点から生成したZIF-8の結晶性は高いと言える。
メカノケミカル法の特徴のもう一つは,出発原料の酸化亜鉛の粒子径を変えることにより,コアシェル型の複合微粒子が得られるところにある(Fig. 4b)。メカノケミカル法を利用したZIF合成は,Friščićらが先駆けて報告している12)。しかし,Friščićらはneatでの酸化亜鉛とイミダゾール系架橋配位子の反応は進行しないと結論しており,ZIF生成を促進させるためにイオン性物質や有機溶媒を添加するion- and liquid-assisted grinding(ILAG)法を提案している。ではなぜ,筆者らの方法ではneatでの酸化亜鉛とHmimの反応が進行し,ZIF-8が生成したのか? その答えは,出発原料の酸化亜鉛の粒子径にある。平均一次粒子径が400 nm程度の酸化亜鉛を用いた場合,酸化亜鉛のZIF-8への転化率は15%程度に留まり,未反応の酸化亜鉛をコア部に多孔性のZIF-8をシェル部にもつユニークな複合微粒子が得られる。固相反応のメカノケミカル法では,酸化亜鉛の粒子径を減少させて反応界面積を増加させれば,neatでの酸化亜鉛とHmimの反応も進行し(平均一次粒子径20 nmの酸化亜鉛の転化率は80%),ZIF-8によって粒子外表面が被覆されることで転換反応が抑制されるというわけである。酸化亜鉛の粒子径,機械的エネルギー,反応時間などを調整することにより,ZIF-8@ZnO型のコアシェル構造を制御できる。多孔性のZIFと金属酸化物を複合化した機能性触媒として期待できる。
配位結合性のMOF/PCPは物理的・化学的安定性が低いものが多く,分離膜部材として要求される耐久性,製膜性などの基準を満たすMOF/PCPは限定される。CaroらがKnudsen拡散支配を超える性能を示すZIF-8膜を報告して以来,ZIFを利用した分離膜開発が活発化している13)。SODをはじめ,RHO,LTA,MER,ANA,BCT,DFT,GIS,GMEなどゼオライトと同様のトポロジーに加えて,ゼオライトでは報告例のない構造を合わせ,さらに異なる組成も含めてZIFの種類は100を超えるが,膜化された種類はその1割程度である。膜化が検討された報告例は,ZIF-7(SOD; 細孔開口部0.30 nm)14),ZIF-8(SOD; 0.34 nm)13,15),ZIF-22(LTA; 0.30 nm)16),ZIF-69(GME; 0.44 nm)17),ZIF-78(GME; 0.38 nm)18),ZIF-90(SOD; 0.35 nm)19),ZIF-93(RHO; 0.36 nm)20),ZIF-95(POZ; 0.37 nm)21),ZIF-100(MOZ; 0.34 nm)22)などであるが,その報告数の7割をZIF-8が占める。また,分離系は水素分離(H2/N2,H2/CO2,H2/CH4,H2/C2H6,H2/C3H8),燃焼排ガスからの二酸化炭素回収(CO2/N2),天然ガス・バイオガス精製(CO2/CH4),オレフィン/パラフィン分離(C2H2/C2H4,C3H6/C3H8)を対象としている。報告数のうちおよそ5割を水素分離,およそ2割をガス精製が占めており,その後に二酸化炭素回収,オレフィン/パラフィン分離が続く。しかしながら,水素は他の分子に比べて小さく,拡散係数が大きいため,必ずしも水素分離系が膜性能を判定する指標に適しているわけではない。また,ずっと安価な高分子膜が商業利用されている中,新規参入のZIF,MOF/PCPは水素分離よりもオレフィン/パラフィン分離への展開が期待されている23)。
ZIFはその構造,形態そのものがゼオライトと類似していることから,ZIF製膜の研究は先行のゼオライト膜合成に倣ったものが多い。また,他の無機膜と同様に膜の機械的強度を確保するために,多孔質アルミナなどを支持体として,その上に製膜されることが多い。一方,ゼオライト合成とは異なり,構造規定剤が不要であること,常温・常圧の温和な条件で合成できることがZIFの製膜に活かされている。また,ZIF合成プロセスの特徴を活かして,ゼオライト膜合成にはないユニークな製膜手法(contra-diffusion24)やelectrochemical deposition25),layer-by-layer26))も報告されている。ここでは,水系合成によるZIF-8の製膜に焦点を絞って解説する。
ゼオライトとZIFはその構造と結晶性の形態の特徴を共有しているものの,その結合様式には共有結合と配位結合の大きな相違点がある。また,ゼオライト膜合成における結晶の溶解・再析出による膜成長を促すほどの過酷な条件(酸・アルカリ性,高温高圧)をZIF合成では必要としない。にもかかわらず,ZIFの製膜はゼオライト膜合成と同様,核となる種結晶を使用し,それを二次成長させて製膜する傾向にある。MOF/PCPの製膜手法として,種結晶を用いる代わりに支持体を3-aminopropyltriethoxysilane(APTES)で表面修飾してin situで核生成・膜成長させる方法19,21)やMOF/PCPを構成する金属種を支持体として直接結晶層を形成させる方法27)がある。このようなin situ法は,APTESあるいは支持体の金属種がアンカーとして結晶/支持体間の接合性を担保する手段としても理にかなっている。しかしながら,水系合成法は反応溶液の反応性が極めて高いため,種結晶フリーなin situ法において支持体上で不均一核生成を促し,膜成長させることは困難とされてきた28)。
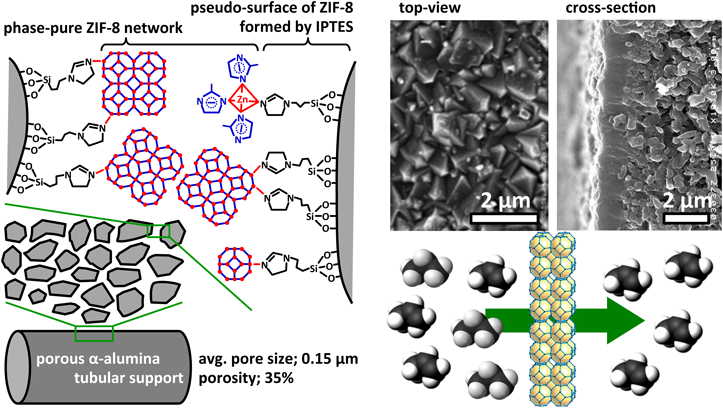
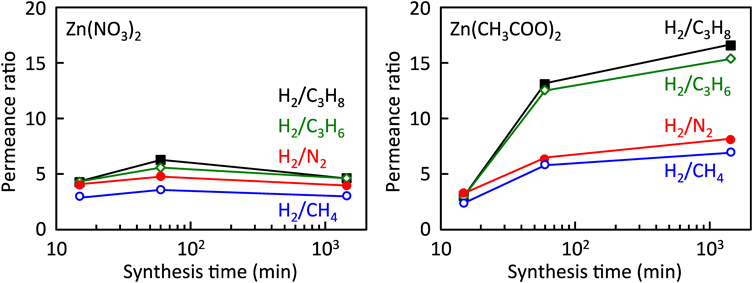
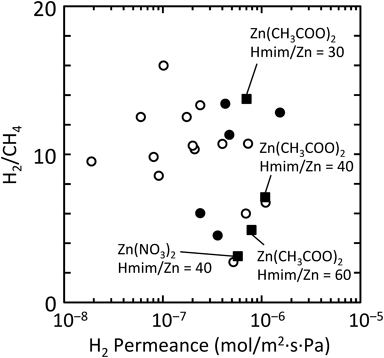
そこで,筆者らは水系合成によるZIF-8生成をin situでの製膜法に適用すべく,多孔質アルミナ支持体の修飾とZIF-8生成速度について検討した。イミダゾール基をもつシリル化剤3-(2-imidazolin-1-yl)propyltriethoxysilane(IPTES)を用いて,多孔質アルミナ支持体をZIF-8粒子表面の末端構造に模することにより,支持体表面上にZIF-8を不均一核生成させ,優先的に膜成長させる条件を探索した(Fig. 5)29,30)。硝酸亜鉛を用いた場合,水溶媒中でのZIF-8の生成速度は極めて速く,10秒程度で製膜溶液は白濁する。透過率比が合成時間に依存せずほぼ一定であることから,膜の緻密化が進行していないことがわかる(Fig. 6)。一方,酢酸亜鉛を用いることにより,合成時間とともに透過率比が向上した。酢酸亜鉛はキレート構造を有する亜鉛源であり,酢酸イオンとHmimとの配位交換による反応速度の低下により,反応溶液バルク側での核生成の抑制と支持体上での優先的な結晶成長が促進され,緻密膜が形成したと考えられる。また,酢酸亜鉛を用いれば,亜鉛–水分子の配位競合も抑制され,水系合成におけるHmimの過剰添加量を低減した条件においても良好な結晶性を有するZIF-8が生成することが確認された。Hmim/Zn比が低く反応速度の遅い成長溶液を用いるほど透過率比が向上した。Hmim/Zn比の膜厚依存性はないことから,製膜溶液の組成によって気体透過特性が異なることは,製膜速度の違いにより膜のミクロ構造が異なっていることを示唆している。種結晶フリーで水系合成により作製したZIF-8膜は,既報のZIF-8膜と同程度のH2/CH4透過率比をもちながら(実験条件がそれぞれ異なるため,直接的な比較は困難であるものの),比較的高い水素透過性を示すところに特徴がある(Fig. 7)。
3.および4.で紹介したように,筆者らはこれまで「水を溶媒とする反応晶析操作」や「無溶媒のメカノケミカル操作」でZIF-8の結晶品質(晶癖,粒子径分布,結晶多形,高次形態)を制御してきた。水系合成では,亜鉛塩の種類や合成温度,Hmim/Zn比を調整することにより,ZIF-8の粒子径を制御できる。MOF/PCPの応用を検討するための基礎的な測定として吸着測定が挙げられる。PFG-NMR31)やIR Microscopy32)を用いれば,結晶内のゲスト分子の拡散性を測定・解析することができる。近年の測定技術の向上により,小さな結晶であっても測定・解析が可能となってきているが,それでも数十〜数百µm程度の大きさをもつ単結晶が必要であり,吸着挙動に対する粒子径の依存性に関する研究例は未だ少ない。しかし,粒子径や形態の依存性を把握することは,吸着や膜分離への展開を検討する上で重要な課題である。ここでは筆者らが検討してきたN2,Arおよびn-butanolの吸着における粒子径の依存性について解説し33),分離膜の作製指針と今後の課題について整理する。
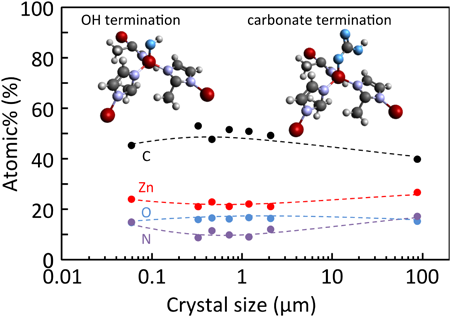
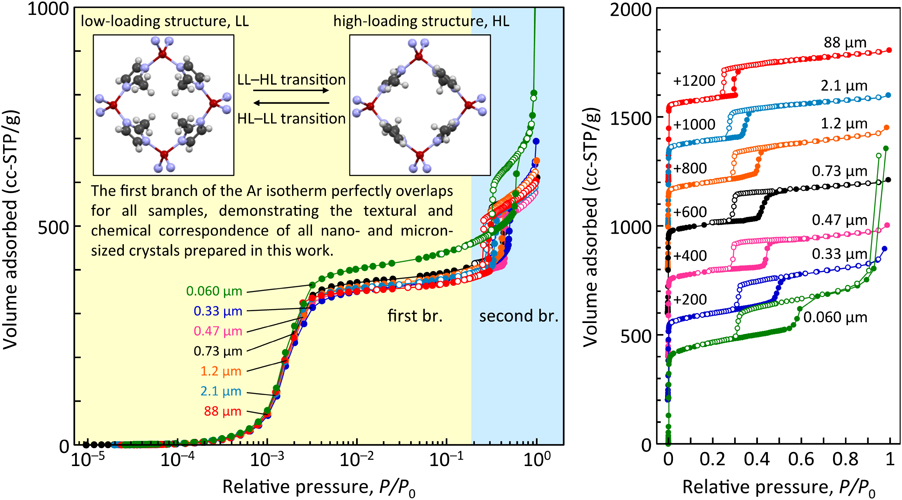
亜鉛源,Hmim/Zn比,溶媒,添加物などの合成条件を変化させ,ZIF-8の平均粒子径を0.060から88 µmまで制御した。比表面積と細孔容積は粒子径に依存しないことが確認された。また,XRD,Raman,XPS測定から評価する限りでは,構造および化学的性質にも粒子径の依存性は認められなかった。一方,XPS測定によるZIF-8粒子(化学量論はC8H10N4Zn)の最表面近傍の組成分析から,末端には水酸基や水酸基に大気中の二酸化炭素が反応したカルボネート基が存在することが示唆された(Fig. 8)。N2およびAr吸着等温線において,それぞれ相対圧0.005〜0.02および0.3〜0.6付近にステップ状の吸着量増加が確認された(Fig. 9にAr吸着等温線をログスケールおよびリニアスケールで示す)。また,そのステップ状の吸着量増加は粒子の微細化とともに高圧側にシフトしていることがわかる。MOF/PCPはゲスト分子の吸着に誘起されて,架橋配位子を回転させ構造を柔軟に変化させるゲートオープニング機能を有することが知られている。架橋配位子が回転し,ケージ状のナノ空間へのゲートが閉じた状態から開いた状態へ変化するにはある閾圧力(ゲート圧)が必要であり,このゲート圧はゲスト分子の種類および架橋配位子の種類によって異なる。ZIF-8のN2およびAr吸着に見られるステップ吸着挙動は,low-loading(LL)構造からhigh-loading(HL)構造への吸着誘起構造転移によるものであると理解されている34)。ログスケールで示した吸着等温線から,LLならびにHL構造におけるArの吸着量には粒子径依存性は認められず(0.060 µmの粒子には粒子間隙による毛管凝縮が観察されるものの),0.060から88 µmまで粒子径制御したZIF-8粒子はどれも良好な細孔構造,結晶性をもつことがわかる。粒子の微細化による構造転移圧の高圧化を示す結果は,ZIF-8の粒子径により細孔空間機能を制御できることを示唆する。LL–HLの構造転移圧力を構造転移の難易とし,構造を「かたい」,「やわらかい」と仮に表現するならば,ZIF-8の粒子径を制御することにより,構造の「かたさ」,「やわらかさ」を微調整できるということになる。[Cu2(dicarboxylate)2(amine)]nによるメタノール吸着においても同様に,粒子径を小さくすることにより吸着圧が高圧側へシフトすることが報告されており,粒子の微細化により構造柔軟性が喪失すると考察されている35)。ZIF-8の細孔開口部は0.34 nmであるが,0.76 nmの1,2,4-trimethylbenzeneまでも吸着されることが知られており36),構造柔軟性があるがゆえにシャープな分子ふるい分画範囲の欠如が認められる。ZIF-8の構造柔軟性が粒子径によって変化するならば,粒子径の制御による分子ふるい効果の調節が期待できる。
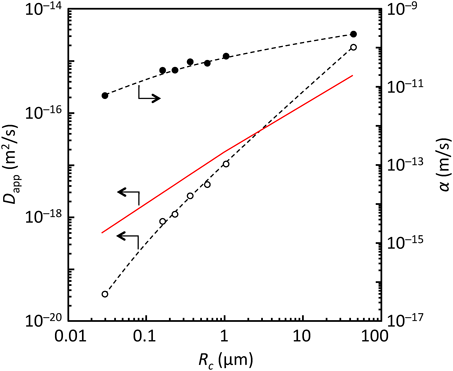
一方,ZIF-8によるn-butanol吸着には,[Cu2(dicarboxylate)2(amine)]nによるメタノール吸着に見られるゲート圧の粒子径依存性は確認されなかった。ZIF-8の粒子径の減少とともに吸着速度が速くなるのは妥当な結果であるが,微細化ほどに吸着速度は大きくならず,粒内拡散機構(eq. 4)から外れることが確認され,表面バリア層(eq. 5)の物質移動抵抗の存在が示唆された。また,ZIF-8の微細化による表面抵抗の寄与の増大に加え,粒子径が小さくなるとともに表面透過係数そのものが減少することが示唆された(Fig. 10)。粒内拡散と表面抵抗の相対的な寄与度と表面透過係数の粒子径依存の両方が吸着速度を支配していると考えられる(eq. 6)。吸着平衡と吸着速度を同じ土俵で議論することは危険であり,より詳細な検討を進める必要があるが,吸着速度の粒子径依存性の一因として粒子径で変化する構造柔軟性の影響が考えられる。粒子径によって吸着特性が変化することは,多結晶膜の結晶粒の精密制御により分離性能の調整が可能であることを意味している。もちろん,多結晶膜において粒子径を変化させるということは,粒界の面積,構造の変化も付随することになり,それぞれの影響を抽出して議論することは難しいわけであるが。
ゼオライト膜と同様の多結晶膜の開発と並行して,MOF/PCPと高分子材料を複合化したMixed Matrix Membrane(MMM)の研究37)も活発化しており,より一層無機と有機の境界領域に踏み込んだ研究分野として発展しつつある。設計自由度の高さと構造柔軟性という魅力をもつMOF/PCPに期待されるところは多いが,分子ふるい機構による吸着・膜分離において「構造柔軟性」は果たして利点となりうるのかどうか,現時点で結論を出すのは難しい。構造の膨張・収縮や細孔開口部の配位子の運動による細孔径の変化のために天然ガス重質分を吸着する一方,脱離させにくいという課題や,既報の測定条件の〜3 bar程度の膜間差圧による分離性能が実プロセスの高圧条件でも担保されるのかという課題など検討事項は多い。また,原料ガスに同伴しうる炭酸ガスや硫化水素など水溶性の腐食性成分,酸性ガス成分はたとえ微量であるとしてもMOF/PCPの劣化を招く恐れがあり,長期運用での膜の耐久性試験は今後の検討課題である。さらに,脱水縮合しうる表面末端水酸基をもつゼオライトに比べて,架橋配位子を表面末端とするMOF/PCPの結晶粒界の緻密化はより高い課題意識で取り組むべきだろう。
本稿では,より一層の簡便性・量的生産性を図るために筆者らが取り組んできた「水系合成法」および「メカノケミカル法」によるZIF-8の結晶品質(晶癖,粒子径分布,結晶多形,高次形態)の制御を中心に紹介し,膜形成への展開を解説した。多結晶型の分離膜を主に紹介したが,MOF/PCPと高分子を複合化したMMMの研究開発も盛んであり,無機と有機の境界領域にまたがって研究領域が急速に拡大・深化している。MOF/PCPが多孔性材料として見出されて20年近くになるが,工業的応用を見据えて,精密な構造設計に加えて,長期安定性・耐久性に関する知見ならびに環境・コスト面でも有望な製造技術の体系化が今後ますます重要になる。また,MOF/PCPが先行のゼオライトなど既存の多孔性材料に対して工業的優位性で肩を並べる影響力をもつためには,MOF/PCPの特徴を活かした独自の用途分野を展開していくことが望まれる。
謝辞Acknowledgments
本研究の一部は,JSPS科研費25790019,文部科学省私立大学戦略的研究基盤形成支援事業,(公財)関西エネルギー・リサイクル科学研究振興財団の助成を受けて行いました。また,平成26年度関西大学在外研究による成果を含みます。在外研究を快く承諾して頂いたVrije Universiteit BrusselのJoeri F. M. Denayer教授に深謝いたします。本研究は,来田康司博士(現:RITE研究員)をはじめとする本学学生ならびに多くの共同研究者によって遂行されました。ここに感謝の意を表します。
引用文献References
1) O. M. Yaghi, M. O' Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi, J. Kim, Nature, 423, 705–714 (2003); S. Kitagawa, R. Kitaura, S. Noro, Angew. Chem. Int. Ed., 43, 2334–2375 (2004); G. Ferey, Chem. Soc. Rev., 37, 191–214 (2008); J. R. Li, R. J. Kuppler, H. C. Zhou, Chem. Soc. Rev., 38, 1477–1504 (2009); M. Shah, M. C. McCarthy, S. Sachdeva, A. K. Lee, H. K. Jeong, Ind. Eng. Chem. Res., 51, 2179–2199 (2012); S. L. Qiu, M. Xue, G. S. Zhu, Chem. Soc. Rev., 43, 6116–6140 (2014).
2) K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe, O. M. Yaghi, Proc. Natl. Acad. Sci. USA, 103, 10186–10191 (2006).
3) J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem, R. R. Willis, J. Am. Chem. Soc., 131, 15834–15842 (2009).
4) S. Tanaka, K. Kida, M. Okita, Y. Ito, Y. Miyake, Chem. Lett., 41, 1337–1339 (2012).
5) K. Kida, M. Okita, K. Fujita, S. Tanaka, Y. Miyake, CrystEngComm, 15, 1794–1801 (2013).
6) J. Cravillon, C. A. Schröder, H. Bux, A. Rothkirch, J. Caro, M. Wiebcke, CrystEngComm, 14, 492–498 (2012).
7) S. Tanaka, K. Kida, T. Nagaoka, T. Ota, Y. Miyake, Chem. Commun., 49, 7884–7886 (2013).
8) 齋藤文良,粉砕,39, 24–38 (1995); 51, 24–29 (2008).
9) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steedk, D. C. Waddelli, Chem. Soc. Rev., 41, 413–447 (2012).
10) K. Yamamoto, S. E. B. Garcia, F. Saito, A. Muramatsu, Chem. Lett., 35, 570–571 (2006).
11) T. Wakihara, J. Tatami, J. Jpn. Petrol. Inst., 56, 206–213 (2013).
12) P. J. Beldon, L. Fábián, R. S. Stein, A. Thirumurugan, A. K. Cheetham, T. Friščić, Angew. Chem. Int. Ed., 49, 9640–9643 (2010); M. J. Cliffe, C. Mottillo, R. S. Stein, D.-K. Bučarb, T. Friščić, Chem. Sci., 3, 2495–2500 (2012).
13) H. Bux, F. Y. Liang, Y. S. Li, J. Cravillon, M. Wiebcke, J. Caro, J. Am. Chem. Soc., 131, 16000–16001 (2009).
14) Y. Li, F. Liang, H. Bux, W. Yang, J. Caro, J. Membr. Sci., 354, 48–54 (2010); T. Li, Y. Pan, K.-V. Peinemann, Z. Lai, J. Membr. Sci., 425–426, 235–242 (2013).
15) H. Bux, C. Chmelik, R. Krishna, J. Caro, J. Membr. Sci., 369, 284–289 (2011); M. Askari, T. S. Chung, J. Membr. Sci., 444, 173–183 (2013); A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones, S. Nair, Science, 345, 72–75 (2014); H. T. Kwon, H.-K. Jeong, Chem. Eng. Sci., 124, 20–26 (2015).
16) A. Huang, H. Bux, F. Steinbach, J. Caro, Angew. Chem., 122, 5078–5081 (2010).
17) Y. Liu, E. Hu, E. A. Khan, Z. Lai, J. Membr. Sci., 353, 36–40 (2010).
18) X. Dong, K. Huang, S. Liu, R. Ren, W. Jin, Y. S. Lin, J. Mater. Chem., 22, 19222–19227 (2012).
19) A. Huang, N. Wang, C. Kong, J. Caro, Angew. Chem., Int. Ed., 51, 10551–10555 (2012); A. Huang, W. Dou, J. Caro, J. Am. Chem. Soc., 132, 15562–15564 (2010).
20) F. Cacho-Bailo, G. Caro, M. Etxeberria-Benavides, O. Karvan, C. Tellez, J. Coronas, Chem. Commun., 51, 11283–11285 (2015).
21) A. Huang, Y. Chen, N. Wang, Z. Hu, J. Jiang, J. Caro, Chem. Commun., 48, 10981–10983 (2012).
22) N. Y. Wang, Y. Liu, Z. W. Qiao, L. Diestel, J. Zhou, A. S. Huang, J. Caro, J. Mater. Chem. A, 3, 4722–4728 (2015).
23) C. Zhang, W. J. Koros, J. Phys. Chem. Lett., 6, 3841–3849 (2015).
24) J. Yao, D. Dong, D. Li, L. He, G. Xu, H. Wang, Chem. Commun., 47, 2559–2561 (2011); N. Hara, M. Yoshimune, H. Negishi, K. Haraya, S. Hara, T. Yamaguchi, J. Membr. Sci., 450, 215–223 (2014).
25) A. M. Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn, J. Gascon, Cryst. Growth Des., 12, 3489–3498 (2012).
26) G. Lu, J. T. Hupp, J. Am. Chem. Soc., 132, 7832–7833 (2010).
27) H. Guo, G. Zhu, I. J. Hewitt, S. Qiu, J. Am. Chem. Soc., 131, 1646–1647 (2009); Y. Hu, X. Dong, J. Nan, W. Jin, X. Ren, N. Xu, Y. M. Lee, Chem. Commun., 47, 737–739 (2011); Z. Kang, M. Xue, L. Fan, J. Ding, L. Guo, L. Gao, S. Qiu, Chem. Commun., 49, 10569–10571 (2013).
28) Y. C. Pan, Z. P. Lai, Chem. Commun., 47, 10275–10277 (2011); Y. C. Pan, T. Li, G. Lestari, Z. P. Lai, J. Membr. Sci., 390–391, 93–98 (2012); Y. C. Pan, B. Wang, Z. P. Lai, J. Membr. Sci., 421–422, 292–298 (2012).
29) K. Kida, K. Fujita, T. Shimada, S. Tanaka, Y. Miyake, Dalton Trans., 42, 11128–11135 (2013).
30) S. Tanaka, T. Shimada, K. Fujita, Y. Miyake, K. Kida, K. Yogo, J. F. M. Denayer, M. Sugita, T. Takewaki, J. Membr. Sci., 472, 29–38 (2014).
31) A. K. Pusch, T. Splith, L. Moschkowitz, S. Karmakar, R. Biniwale, M. Sant, G. B. Suffritti, P. Demontis, J. Cravillon, E. Pantatosaki, F. Stallmach, Adsorption, 18, 359–366 (2012).
32) H. Bux, C. Chmelik, J. M. van Baten, R. Krishna, J. Caro, Adv. Mater., 22, 4741–4743 (2010).
33) S. Tanaka, K. Fujita, Y. Miyake, M. Miyamoto, Y. Hasegawa, T. Makino, S. Van der Perre, J. C. S. Remi, T. Van Assche, G. V. Baron, J. F. M. Denayer, J. Phys. Chem. C, 119, 28430–28439 (2015).
34) D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons, T. Düren, J. Am. Chem. Soc., 133, 8900–8902 (2011); C. O. Ania, E. García-Peŕez, M. Haro, J. J. Gutieŕrez-Sevillano, T. Valdeś-Solís, J. B. Parra, S. Calero, J. Phys. Chem. Lett., 3, 1159–1164 (2012); L. Zhang, Z. Hu, J. Jiang, J. Am. Chem. Soc., 135, 3722–3728 (2013); H. Tanaka, S. Ohsaki, S. Hiraide, D. Yamamoto, S. Watanabe, M. T. Miyahara, J. Phys. Chem. C, 118, 8445–8454 (2014).
35) Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata, S. Kitagawa, Science, 339, 193–196 (2013).
36) K. Zhang, R. P. Lively, C. Zhang, R. R. Chance, W. J. Koros, D. S. Sholl, S. Nair, J. Phys. Chem. Lett., 4, 3618–3622 (2013).
37) M. J. C. Ordonez, K. J. Balkus, J. P. Ferraris, I. H. Musselman, J. Membr. Sci., 361, 28–37 (2010); T. H. Bae, J. S. Lee, W. L. Qiu, W. J. Koros, C. W. Jones, S. Nair, Angew. Chem. Int. Ed., 49, 9863–9866 (2010); J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastian, C. Casado, J. Coronas, Chem. Mater., 24, 2829–2844 (2012); B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn, J. Gascon, Chem. Soc. Rev., 44, 2421–2454 (2015).