層状ケイ酸塩は架橋水酸基構造を持つアニオン性シリケートシートと層間の交換性カチオンから成る結晶性の層状化合物の一種である。ケイ素を基礎構成元素とする他のナノ空間材料(例えば,ゼオライト,メソポーラスシリカ)が剛直な3次元の骨格構造を有しているのに対して,2次元構造を有する層状ケイ酸塩は,様々な構造変換を可能とする柔軟な修飾性と機械的,熱的に安定な結晶性骨格の寄与によって,多彩かつ合理的な機能設計が可能となる。
層状ケイ酸塩層表面に存在する架橋水酸基(SiOHとSiO−基のペア)はアルコールや有機シランに代表されるゲスト分子と共有結合を形成することが可能であり,空間的に固定されたゲスト種の寄与によって特異的な機能(例えば分子認識的な吸着作用)の発現が可能となる1,2)。一方で,層状ケイ酸塩のシリケート骨格構造がゼオライトと高い構造類似性を持つ事実から,架橋水酸基の脱水縮合(幾何学的固相転換:Topotactic conversion)を経由した層状ケイ酸塩からのゼオライト合成に関する研究も精力的に行われてきた。MWWやFERが層状前駆体を経由して得られるゼオライトであることはそれぞれ1994および1996年に報告されたが3,4),層状ケイ酸塩のみから得られる新規のゼオライト(CDO5), NSI6), RWR7), RRO8))が報告されたのを皮切りに,層状ケイ酸塩からのゼオライト構築およびその転換過程は詳細に検討され,現在では10種以上のゼオライト骨格が層状構造から合成可能である。
また,層状ケイ酸塩の層間水酸基に対してシリル化剤を作用させることで(層間シリル化)層間に規則的に四面体ピラーを配置した新規細孔構造も構築可能であり,稲垣9a)や池田9b)らの研究が先駆けとなり,これらゼオライト様新規化合物の設計も盛んに行われている。最近では,Čejkaらの研究グループがUTLゼオライト骨格中のゲルマニウムのみを選択的に取り除くことで,2次元構造を持った構造体(IPC-1P)への転換を報告し,そこからさまざまな新規構造群の設計に成功している9c)。一方で,Ryooらが合成に成功した層状構造を持ったMFIゼオライトは,ゼオライトの機能設計における2次元構造の重要性を再確認させた10)。このような層状ケイ酸塩および層状ゼオライト前駆体を用いた材料設計の可能性や多様性を鑑みると,新規な構造を持つ層状ケイ酸塩の合成は,それらを活用した材料設計の出発地であり,機能設計を拡大させていくために最も有効なファクターであるといえる。
この観点から,我々は新規層状ケイ酸塩の合成を精力的に行い,その結果,様々な構造を持つ層状ケイ酸塩群Hiroshima University Silicates(HUSs)の合成に成功した11)。本解説ではこれらHUSsの合成,基礎物性(吸着特性)評価および機能材料前駆体としての応用例を紹介する。
2. 新規層状ケイ酸塩Hiroshima University Silicate(HUSs)の合成とその構造
層状ケイ酸塩合成はゼオライト合成と類似しており,様々なカチオン源を構造規定剤(structure-directing agent, SDA)として用いた水熱合成によって成される。カチオン源としては無機のアルカリ金属やゼオライト合成で用いられる構造規定剤(4級アンモニウム塩や環状アミン)が用いられる。しかしなら,カチオンの構造と得られるケイ酸塩骨格の因果関係はゼオライト合成にもまして明確でなく,例えば,FERゼオライト前駆体は異なるカチオン源(カリウム,テトラメチルアンモニウム,コリン等)からの合成が報告されている。
我々は層状ケイ酸塩骨格形成の必要条件が構造規定剤の構造でなく水熱合成の別のパラメータにあると考え,水性ゲル中の水量(H2O/SiO2比)に着目した。通常ゼオライトを含む無機化合物の水熱合成はゲルの均一性を保つため,多量の水存在下で行われる(H2O/SiO2比30–50)。一方で,一部の層状ケイ酸塩は極めて少ない水量での合成が報告されており,このパラメータが層状ケイ酸塩を合成するうえで重要なファクターであることが予見された。そこで,極めて水が少ない系(H2O/SiO2比5.5の条件)で様々な構造を持つ4級アンモニウムカチオンを構造規定剤として用いて層状ケイ酸塩の合成を試みた。
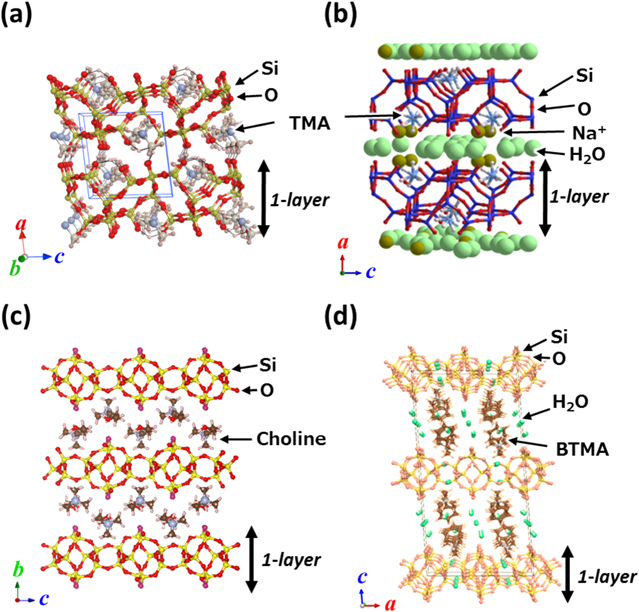
Table 1に得られた層状ケイ酸塩HUSsの合成条件および構造的に関連する(構造類似性があるまたは幾何学的固相転換が可能な)ゼオライトの構造を示す。Figure 1にHUSsの結晶構造を示す。出発水性ゲルのH2O/SiO2比のみを変化させて合成を行った場合,いずれの構造規定剤を用いた条件でも多量の水存在下では(H2O/SiO2比30–50),ゲル中のシリカの溶解性が高まり生成物は得られなかった。このことは低いH2O/SiO2比という水熱合成条件が層状ケイ酸塩HUSsを得るために極めて重要なファクターであることを示している。
Table 1. Synthesis conditions of layered silicates HUSs| Layered silicate | SDA cation | OSDA/SiO2 ratio | NaOH/SiO2 ratio | H2O/SiO2 ratio | Related zeolite type |
| HUS-1 | Tetramethylammonium | 0.6 | 0.2 | 5.5 | AST, SOD |
| HUS-5 | Tetramethylammonium | 0.6 | 0.2 | 5.5 | AST, SOD |
| HUS-2 | Choline | 0.4 | 0.2 | 5.5 | HEU |
| HUS-3 | Choline | 0.2 | 0.2 | 5.5 | CDO |
| HUS-4 | Choline | 0.6 | 0.2 | 5.5 | CDO |
| HUS-7 | Benzyltrimethylammonium | 0.2 | 0.2 | 5.5 | HEU |
テトラメチルアンモニウム(TMA)カチオンを用いて得られた層状ケイ酸塩HUS-111a)とHUS-511e)はSODゼオライト骨格を分割したような同様のシリケート骨格構造を有するが,HUS-5は水分子とナトリウムカチオンが層間に存在することでより広い層間距離を持つ。HUS-5は既存の層状ケイ酸塩β-HLS12)と同様の結晶相であったが,HUS-1は層間のカチオンおよびシリケートシートの積層規則が異なる新規な結晶構造を有する。この2種の層状ケイ酸塩は可逆的に結晶相の構造変換が可能であり,例えば,HUS-1を水酸化ナトリウム水溶液で処理すると層間にナトリウムカチオンが挿入されHUS-5が得られ,一方で,HUS-5をpH 7以下の水溶液で洗浄することで層間からナトリウムカチオンが放出されHUS-1へと転換する。
コリンカチオンを用いた水熱合成によって,層状ケイ酸塩HUS-2,HUS-3,およびHUS-4が得られた。HUS-3およびHUS-4はその結晶構造を明らかにすることはできなかったが,空気下で350°Cの焼成処理を行うことによってCDOゼオライトへと構造転換が進行したことから,PREFERやPLS-1などと類似したシリケート骨格構造を持つと予想された。一方で,MAS NMRおよび粉末X線回折を用いたリートベルト解析によって,層状ケイ酸塩HUS-2の結晶構造解析には成功し,HUS-2は既存にはない新規なシリケート骨格構造を持つことを明らかにした。HUS-2の骨格構造はbre(10T)型のコンポジットビルディングユニットを持つHEUゼオライトと酷似しており,4-,5-,6-員環構造をシリケートシート内部に有する。また,層間のコリンカチオンを電荷補償している架橋水酸基は結晶格子のa軸方向に向かって平行に結合方向を向けて配列しており,シリケートシート間で水酸基が架橋する層状ケイ酸塩PLS-15),水酸基の向きが層表面で交差しているHUS-1など他の層状ケイ酸塩と比べても特異なことがわかる11b)。
ベンジルトリメチルアンモニウム(BTMA)カチオンを構造規定剤として用いた場合は層状ケイ酸塩HUS-7が得られる。構造規定剤の構造や大きさが異なるにもかかわらずHUS-7はHUS-2と同様のシリケート骨格を持っており,このことからもゼオライト合成に比べて,層状ケイ酸塩の骨格構造形成に構造規定剤の構造の寄与が少ないことが伺える。ただしHUS-2とHUS-7の結晶構造は大きく異なり,HUS-2のシリケートシートが比較的狭い距離で積層しているのに対し,HUS-7の基本面間隔は約1.7 nmと非常に広く,さらに層間の有機カチオンは層状ケイ酸塩RUB-5113)に見られるようなベンゼン環同士のパッキングによって配列しているのではなく,層間の水和水とともに結晶格子のb軸方向に向かって複数のBTMAカチオンが集合した状態で配列している。
3.1 HUS-1を用いた海水からのNi2+の濃集11f)
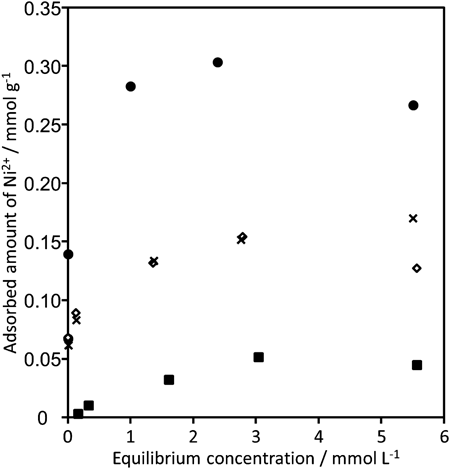
井出らは天然にも存在する層状ケイ酸塩(層状アルカリシリケート)であるマガディアイトが海水からZn2+イオンを濃集することを報告した14)。マガディアイトは選択的かつ効率的にZn2+イオン吸着したが,異なるシリケート骨格構造を持つアルカリシリケート(オクトシリケート)はほとんどZn2+を吸着せず,層状ケイ酸塩の骨格構造の違いが特定のイオンの選択的吸着を進行させていることが予想された。そこで,HUS-1を用いて海水からのNi2+イオンの濃集を試みた。また,層状ケイ酸塩(マガディアイト),粘土鉱物(Na型モンモリロナイト),MORゼオライトも比較吸着剤として用い,吸着実験にはNi2+イオンを含有(10–300 ppm)した海水の組成を模した電解質溶液を用いた。Figure 2に電解質溶液からのNi2+の吸着等温線を示す。Na+,K+,Mg2+,およびCa2+などの他のカチオン濃度がNi2+の濃度と比べて非常に高いもかかわらず(例えばNa+は約4000 ppm),HUS-1のNi吸着等温線は低濃度で急激な吸着量の増加が観察されるH-型15)であり,これはNi2+とHUS-1の間の強い相互作用を示している。また,吸着したカチオン組成を調査したところ,HUS-1からはNi2+以外の元素はほとんど検出されず(吸着したカチオンの約90%がNi2+),選択的かつ効率的なNi2+の吸着が進行していることが確認できた。一方で,他の吸着剤には一定量のNi2+の吸着は観察されたものの多量の海水含有成分の存在が観察され,HUS-1のNi2+濃縮材料としての有用性が示された。
TG-DTAによって見積もった吸着前後で脱離した層間カチオン量,吸着前後のXRD,29Si MASNMR,SEM/EDXマッピングの結果から,HUS-1へのNi2+の吸着は層間のTMAとのイオン交換によって進行し,さらにイオン交換中HUS-1の結晶構造にほとんど変化がないことが分かった(シリケートシートがイオン交換によって拡張していない)。海水に含まれているイオンの大きさと,水和エネルギーから(イオン直径:Na+=0.19 nm,K+=0.26 nm,Mg2+=0.13 nm,Ca2+=0.20 nm,Ni2+=0.14 nm,水和エネルギー:Na+=114 kcal g−1 ion−1,K+=94 kcal g−1 ion−1,Mg2+=490 kcal g−1 ion−1,Ca2+=410 kcal g−1 ion−1,Ni2+=516 kcal g−1 ion−1),Ni2+は比較的小さいイオンであり,なおかつ非常に高い水和エネルギーを持っていることが分かる。また,HUS-1にはNi2+と同程度のイオン直径,水和エネルギーを持つMg2+も比較的選択的に吸着したことから,HUS-1の非常に狭い層間距離(0.15 nm)が,小さなイオンであるNi2+の選択的な吸着を促したと考察した。
3.2 HUS-7の可逆的かつ選択的なフェノール吸着挙動11g)
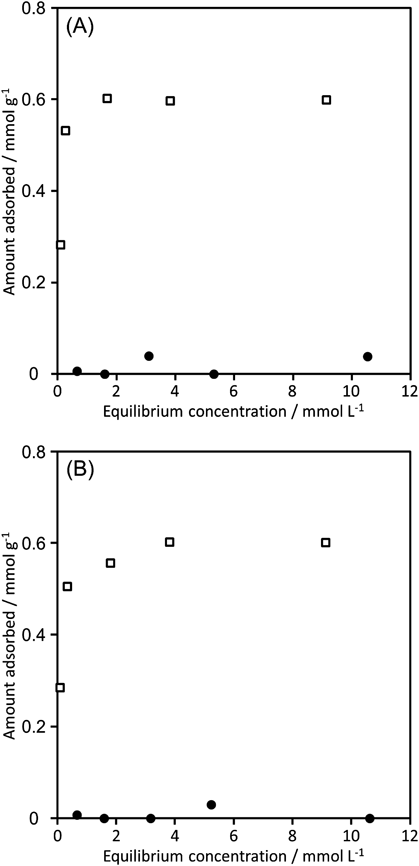
層状ケイ酸塩HUS-7の層間にはBTMAカチオンの集合体からなるピラーと層表面の架橋水酸基が交互に配列している(Figure 1d)。小川らは層状ケイ酸塩層間に2種類の親和性の異なるゲスト種が存在した際の選択的吸着挙動を報告した。彼らは,層状ケイ酸塩層間に空間的に固定された機能ユニットと吸着分子の間に相互的な吸着作用が生じることで選択的吸着特性が生じると結論づけている。このような観点から,HUS-7層間の特異的な分子配列が有機分子の吸着に与える影響を調査した。吸着させる有機分子にはベンゼンとフェノールを選択し,アセトニトリル中で混合系および単成分の吸着実験を行った。Figure 3Aにはアセトニトリルからのベンゼンおよびフェノールの単成分の吸着等温線を示す。ベンゼンはHUS-7にいずれの濃度でも吸着しなかったのに対して,フェノールの吸着等温線は低濃度でも多量の吸着を示し,HUS-7は0.60 mmolg−1のフェノール吸着容量を示した。Figure 3Bのフェノール/ベンゼン混合系の吸着結果から明らかなように(フェノール:ベンゼン存在比=1 : 1),単成分と混合成分での吸着挙動に違いは全くなく,混合系においてベンゼンはフェノールの吸着を全く阻害していないことがわかる。吸着後のHUS-7の評価をXRD,MAS NMR,CHN元素分析から行ったところHUS-7は吸着後も層間距離を維持していること,結晶中にフェノールとBTMAカチオンの存在が確認されたことから,フェノールの吸着はBTMA以外の分子,つまり水分子との交換で進行していることが示唆された。
そこで,吸着したフェノールをHUS-7から除去するために様々な溶媒での洗浄を試みた。アセトンやエタノールで洗浄を行った際にはフェノールはHUS-7中に残存したままだったが,水での洗浄によってフェノールの完全な除去が可能であった。水洗前後においても層間距離の変化はないことは確認されたが,高角度領域のXRDパターンは,「フェノールの吸着によってわずかに変化」→「水洗後は吸着前と同様の回折パターン」という挙動を示し,フェノールと水分子の交換によってHUS-7の結晶構造がわずかに変化していることが示唆された。水洗したHUS-7を再度フェノールの吸着に用いたところ,一回目の吸着実験と遜色ないフェノール吸着容量(0.61 mmolg−1)を示した。層間にBTMAカチオンを有するがその配列がHUS-7と異なる層状ケイ酸塩RUB-51にはベンゼンおよびフェノールとも吸着しなかったことを考慮すると(フェノールおよびベンゼンの吸着量はそれぞれ0.08 mmolg−1および0.06 mmolg−1),この特異的なフェノール吸着挙動はBTMAと表面水酸基が空間的に配置されたHUS-7の特徴的な結晶構造に由来していると考えられる。
上述した以外にも,現在までに,HUS-1のシリル化誘導体によるTMAの選択的吸着11c),およびHUS-2のプロピオン酸に対する分子認識的吸着作用11d)など,新規層状ケイ酸塩の選択的吸着剤としての可能性を明らかにしている。
4.1 層状ケイ酸塩HUS-5イオン交換体を前駆体とした新規ナノポーラスシリカHUS-6の合成11e)
層状ケイ酸塩を用いたメソポーラスシリカの合成は黒田らの研究グループによって精力的に行われてきた16)。FSM-16型のメソポーラスシリカは,高pH条件下でアルキルトリメチルアンモニウムカチオンでイオン交換を行った層状ケイ酸塩カネマイトを前駆体として得られる17–19)。一方,同様の層状ケイ酸塩前駆体でも,酸条件で処理を行うことで「シリケートシートの湾曲」→「層表面の水酸基の縮合」という構造転換を介して異なる細孔構造を持つKSW-2が得られる20)。しかし,メソポーラスシリカの前駆体としてカネマイト以外の層状ケイ酸塩が用いられた例は少なく,異なる骨格構造を持つ層状ケイ酸塩からどのような多孔質シリカが得られるかは大変興味深い。
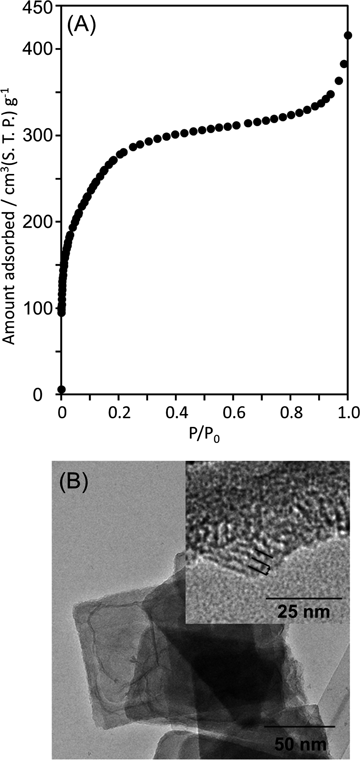
そこで,ポーラスシリカ合成の前駆体としてHUS-1の合成過程で得られた結晶構造の異なる層状ケイ酸塩HUS-5を用いた。HUS-5の結晶構造がHUS-1と異なることは前述したが,HUS-1がその狭い層間距離と層同士の強い相互作用から層間に嵩高いアルキルアンモニウムカチオンを挿入できないのに対して,HUS-5はヘキサデシルトリメチルアンモニウム(C16TMA)カチオンなどの嵩高いアンモニウムカチオンによるイオン交換によって容易な層間拡張が可能である。この層状ケイ酸塩HUS-5のC16TMAイオン交換体に対して様々な後処理を加えることで新規ナノポーラスシリカの合成を試みた。その結果,約1.5 nmの均一な細孔径と約1000 m2 g−1の高い比表面積を持つナノポーラスシリカHUS-6の合成に成功した。Figure 4にHUS-6の窒素吸着等温線およびTEM写真を示す。HUS-6はHUS-5イオン交換体の「酸処理」→「焼成」によって得られる。細孔構造構築過程におけるXRD,13C CP MAS NMR,CHN元素分析結果から,層間の有機物が酸処理後も一部残存していること,さらに29Si MAS NMRから酸および焼成処理によってQ3ピーク強度の著しい低下が観察されたことから,この構造転換は「層状ケイ酸塩のシリケートシートの湾曲」→「層表面間での水酸基の縮合」→「層間のアンモニウムカチオンの燃焼による空孔形成」という過程を経ていると予想された。またHUS-6はX線回折による短周期の規則構造を示さず細孔壁は非晶質だと考えられるが,29Si MAS NMRにはシリケート骨格が極度に開裂したことを示すQ2ピークはほとんど観察されなかった。そのため,構造転換過程でHUS-5のシリケート骨格は局所的には保持されている。また,HUS-6の構造は類似した手法で得られるメソポーラスシリカKSW-2とは細孔径および構造とも大きく異なっているため,カネマイトとHUS-5のシリケートシート構造の差によって異なる細孔構造が得られたと考えられる。
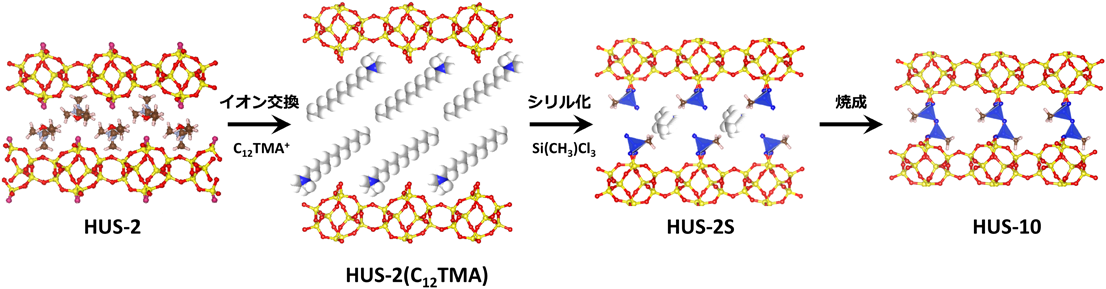
4.2 層状ケイ酸塩HUS-2の層間シリル化によるミクロ多孔質シリカHUS-10の設計11j)
層状ケイ酸塩HUS-2は今までにないシリケート骨格構造を持つ新規の層状ケイ酸塩である。そのため,HUS-2を用いた幾何学的固相転換によるゼオライト化の可能性や層間シリル化によるゼオライト類縁体の設計は興味深い。今のところシリケートの積層規則の最適化や酸処理および焼成条件の詳細な検討を行ったが,HUS-2からゼオライトへの直接的な構造転換は行えていない。しかし,HUS-2へ「イオン交換」→「トリクロロメチルシランによる層間シリル化」→「焼成」という多段階の処理を行うことで規則的ミクロ細孔を有する新規ミクロ多孔質シリカHUS-10の合成に成功した。
Figure 5には層状ケイ酸塩HUS-2からHUS-10への構造転換過程を示す。まず,HUS-2をドデシルトリメチルアンモニウムカチオンでイオン交換した前駆体をトリクロロメチルシランでシリル化する。導入されたシリル化剤由来の四面体ユニットは層表面の水酸基二つと共有結合(Si–O–Si結合)を形成し,層表面の四面体ユニット上にメチル基と,クロロ基の水和の結果生じたシラノール基が残存する。この残存シラノール基を焼成によって脱水縮合させることで,層間で四面体ユニット同士を共有結合(ピラー形成)させ,新規ミクロ多孔質シリカHUS-10を得る。XRD,29Si MAS NMR,CHN元素分析結果より,上述した転換がHUS-2のシリケート骨格構造の崩壊無しに進行していること,ほぼすべての表面水酸基が四面体ユニットで被覆されていることが確認できた。さらに,得られた基礎物性値を元にDFT計算によってHUS-10の構造を予測した。想定されたミクロ多孔質シリカHUS-10の骨格構造は12員環と8員環が交錯した2次元細孔構造を持ち,細孔の交差部分の12員環には四面体ユニット由来のメチル基または焼成時にその加水分解によって生じた水酸基が存在する。
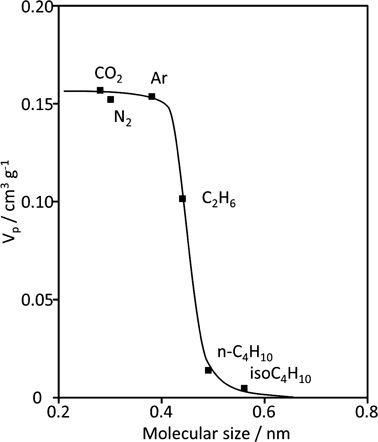
HUS-10の分子ふるい能は分子径の異なるガス成分の吸着等温線を解析することにより確認した。Figure 6にはDA法により各ガスの吸着等温線から得られたミクロ細孔容積と吸着分子の分子径の関係を示す。比較的嵩高い分子(イソブタンやn-ブタン)はほとんど吸着せず,極めて低いミクロ細孔容積しか得られなかった。しかし,エタンから求めたミクロ細孔容積は著しく増加し,さらに,それ以下のサイズの分子の細孔容積は一定の値を示した。このことはHUS-10がエタンより小さな分子を吸着できる分子ふるい能をもつことを示している。すなわち,層状ケイ酸塩HUS-2の層間シリル化を介した精密な構造設計によってゼオライトのような分子ふるい機能を創出できた。
5.1 HUS-2層間へのヘテロポリ酸の挿入による固体酸触媒の調製11h)
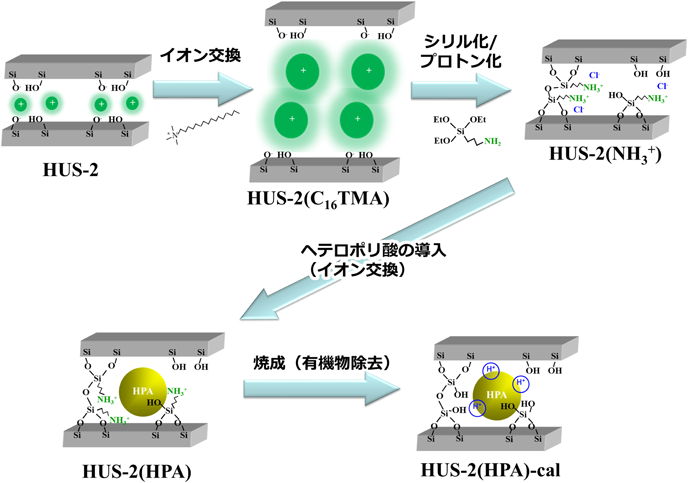
ヘテロポリ酸は強酸性および高い酸化活性を持つ金属酸化物クラスターであり,工業的に有用な触媒である21)。しかし,その極性溶媒への高い溶解性のために触媒反応系からの回収が難しく,高比表面積を有する様々な無機担体へのヘテロポリ酸の担持/導入による不均一系のヘテロポリ酸触媒開発は注目されている。層状ケイ酸塩はその高い耐熱性,耐薬品性および修飾性から触媒担体としても有望であるが,層状ケイ酸塩を用いたヘテロポリ酸含有不均一系触媒の調製はいまだ成されていない。そこで,HUSのうち特に熱安定性の高く,イオン交換による層間の拡張も容易なHUS-2を用い,ヘテロポリ酸を固定した固体酸触媒の設計を試みた。導入するヘテロポリ酸にはKeggin型(H3[PW12O40])およびPreyssler型(H14[NaP5W30O110])のリンタングステン酸を選択した。
Figure 7にはHUS-2層間へのヘテロポリ酸固定化手法を示す。層状ケイ酸塩の骨格はアニオン性である。そのため,ヘテロポリアニオンをイオン交換により導入するために,3-アミノプロピルトリエトキシシランによるシリル化,およびその後のプロトン化処理よって層表面にカチオン性のプロピルアンモニウム基を固定化し,ヘテロポリ酸をイオン交換により導入する。最後に,層間の有機物を焼成によって取り除き,固体酸触媒を調製した。
XRD,MAS NMR,SEM/EDXの結果から,Keggin型ヘテロポリ酸がHUS-2層間に挿入されていることが確認でき,焼成後のBET比表面積およびミクロ細孔容積はそれぞれ206 m2 g−1および0.08 cm3 g−1と,層間のヘテロポリ酸がシリケートシートを支えている多孔質構造を持つことが分かった。一方,Preyssler型ヘテロポリ酸はその熱安定性の低さから,焼成後その構造を安定な状態で保持できなかった。
得られたKeggin型ヘテロポリ酸含有HUS-2の酸触媒特性を流通式の反応装置を用いたエタノールの脱水反応により評価したところ,反応温度300°Cで,約86%のエチレン収率を示し,選択的な脱水触媒として有用であることがわかった。
5.2 層状ケイ酸塩HUS-2への金属アセチルアセチルアセトナートのグラフティングによるチタノシリケート触媒の設計11i)
ゼオライトやメソポーラスシリカの骨格中に4配位状態の遷移金属(チタンやバナジウム)が存在するメタロシリケートはその特異的な触媒活性から有益な選択的酸化触媒として用いられてきた22)。触媒活性点となる4配位遷移金属種の量を増加させることはメタロシリケートの触媒活性を向上させる最も単純な手法である。しかしながら,触媒中の遷移金属種量を顕著に増加させると,選択的な触媒活性を示さない酸化物種(6配位遷移金属種から成るチタニアやバナジア)が必ず生成する。
我々はこのような触媒調製の限界を突破するために,層状ケイ酸塩を活用した触媒設計を試みた。我々は層状ケイ酸塩のポスト処理の一つであるグラフティングに着目した。グラフティングは層状ケイ酸塩ホスト表面へシリル化剤やアルコールなどのゲスト化合物種を共有結合(例えばSi–O–R結合)で固定化する反応である。本研究では触媒設計のホストとして層状ケイ酸塩HUS-2を,固定するゲスト主としてチタニウムアセチルアセトナートを選択し,それらを利用してチタノシリケート触媒を設計した。
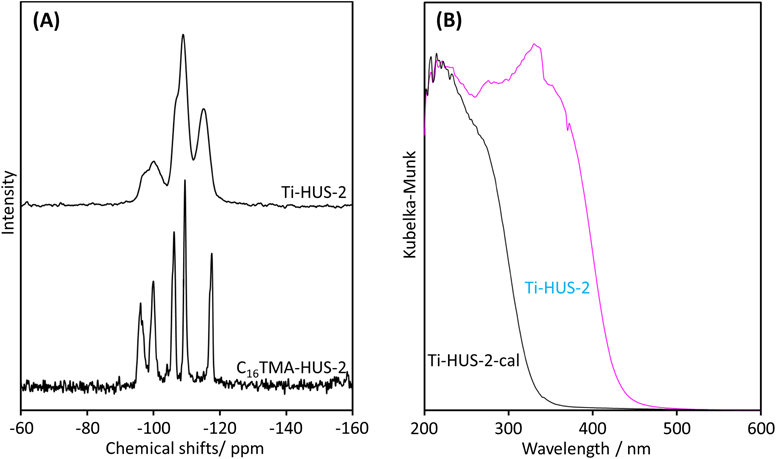
Figure 8にはチタン導入前後の層状ケイ酸塩HUS-2のキャラクタリゼーション結果を示す。
チタニウムアセチルアセトナートの固定化を容易にするため層状ケイ酸塩HUS-2の層間カチオンを嵩高いアルキルアンモニウムカチオン(ヘキサデシルトリメチルアンモニウムカチオン)により交換した(C16TMA-HUS-2)。グラフティング前の29Si MAS NMRには,−100 ppm付近に架橋水酸基に由来するQ3ピークがはっきりと観察されているが,グラフティング処理後(Ti-HUS-2)にこのピークの強度が減少していることがわかる。これは層表面の水酸基とアセチルアセトナートリガンドの縮合反応によって層表面でSi–O–Ti共有結合が形成されたためである(事実グラフティング前後のQ3/Q4ピーク比から表面水酸基の約半分が被覆されていた)。しかし,グラフティング後もUV-Visスペクトルにチタン上のアセチルアセトナートリガンドの存在を示唆する吸収バンドが300–400 nmに観察されたため,層間のチタン種への拡散を容易にするためにサンプルを焼成処理した(Ti-HUS-2-cal)。焼成後サンプルのUV-VisおよびXPSスペクトルおよびTEM観察からはサンプル中でのチタニアの生成はほとんど観察されず,UV-visスペクトルには4配位状態のチタン種に由来する吸収バンドが220 nmにはっきりと観察された。焼成した触媒のチタン含有量は約8 wt%と既存のチタノシリケート触媒(TS-1,約2 wt%)と比較して極めて高かった。
本触媒を溶媒中の飽和酸素を酸化剤とする疑似太陽光照射下のシクロヘキサンの酸化反応に応用した。本反応に対して4配位チタン種を含有するTS-1は選択的にシクロヘキサンの部分酸化物(シクロヘキサノンおよびシクロヘキサノール)を生成した。一方,HUS-2を活用した触媒は,完全酸化物である二酸化炭素をまったく生成せずに,その高いチタン含有量が反映され,TS-1の4倍もの部分酸化物収率を示した。このことから,層状ケイ酸塩を用いた本触媒設計手法が優れたチタノシリケート合成法として極めて有用であることが分かった。また,このチタニウムアセチルアセトナート錯体はメソポーラスシリカチタノシリケート触媒の調製に対しても有用であることも明らかにしている23)。
テトラメチルアンモニウム,コリンおよびベンジルトリメチルアンモニウムカチオンを構造規定剤として用いることで,様々な骨格構造を持つ層状ケイ酸塩HUSsの合成に成功した。
得られたHUSsの吸着剤としての応用を試みたところ,「海水からのNi2+濃縮」,「フェノールの可逆的/選択的吸着」などを含む,他の層状ケイ酸塩および他の材料には成し得ない特異な吸着挙動を発揮した。
HUS-5およびHUS-2を多孔体前駆体として用い,珍しい細孔領域を持つ新規ナノポーラスシリカHUS-6およびゼオライトのような分子ふるい能を持つミクロ多孔質シリカHUS-10の設計/合成に成功した。
HUSsを活用し,酸触媒およびチタノシリケート触媒の設計を試みた。新たな戦略によるヘテロポリ酸固定化触媒およびチタニウムアセチルアセトナート錯体と層状ケイ酸塩の組み合わせによる高チタン含有量を有するチタノシリケートの設計法を提案した。
層状ケイ酸塩はその修飾性に注目するだけでも優れた材料といえるが,それよりも層骨格構造を構造設計の選択肢に入れることができることが,本構造群の最大の利点だと考えている。層状ケイ酸塩層表面の結合可能ポイント(表面水酸基)のバリエーションは層状ケイ酸塩のシリケート骨格構造に由来して任意に変更可能である。今後,安定な骨格と柔軟な修飾性を併せ持つ層状ケイ酸塩のうま味を最大限生かした革新的な材料設計を行っていくためには,材料設計目標に則した最適なシリケートシートの選択が最も重要になるだろう。
謝辞Acknowledgments
本研究を進めるにあたって,以下の方々にご協力頂いた。井出祐介博士(物材機構),佐々木優吉博士(JFCC),吉田要博士(JFCC),関川みゆき博士(JFCC),近江靖則准教授(岐阜大),早川慎二郎教授(広島大),定金正洋准教授(広島大)。中でも,HUSsの構造解析に関しては池田卓史博士(産総研)に多大なご助力を頂いた。ここに記して感謝の意を表する。なお,本研究はJSPS特別研究員奨励費25・1966の助成を受けて行われた。
引用文献References
1) N. Takahashi and K. Kuroda, J. Mater. Chem., 21, 14336 (2011).
2) T. Okada, Y. Ide and M. Ogawa, Chem.–Asian J., 7, 1980 (2012).
3) M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 264, 1910 (1994).
4) L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 6, 259 (1996).
5) T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., 116, 5000 (2004).
6) S. Zanardi, A. Alberti, G. Cruciani and A. Corma, V. Fornes and M. Brunelli, Angew. Chem. Int. Ed., 43, 4933 (2004).
7) B. Marler, N. Stroter and H. Gies, Microporous Mesoporous Mater., 83, 201 (2005).
8) Y. X. Wang, H. Gies, B. Marler and U. Muller, Chem. Mater., 17, 43 (2005).
9) (a) S. Inagaki, T. Yokoi, Y. Kubota and T. Tatsumi, Chem. Commun., 5188 (2007); (b) T. Ikeda, S. Kayamori, Y. Oumi and F. Mizukami, J. Phys. Chem. C, 114, 3466 (2010); (c) W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Čejka, J. Nat. Chem., 5, 628 (2013).
10) M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo Nature, 461, 246 (2009).
11) (a) T. Ikeda, Y. Oumi, K. Honda, T. Sano, K. Momma and F. Izumi, Inorg. Chem., 50, 2294 (2011); (b) N. Tsunoji, T. Ikeda, Y. Ide, M. Sadakane and T. Sano, J. Mater. Chem., 22, 13682 (2012); (c) Y. Ide, M. Torii, N. Tsunoji, M. Sadakane and T. Sano, Chem. Commun., 48, 7073 (2012); (d) N. Tsunoji, Y. Ide, M. Torii, M. Sadakane and T. Sano, Chem. Lett., 42, 244 (2013); (e) N. Tsunoji, M. Fukuda, K. Yoshida, Y. Sasaki, T. Ikeda, Y. Ide, M. Sadakane and T. Sano, J. Mater. Chem. A, 1, 9680 (2013); (f) K. Honda, Y. Ide, N. Tsunoji, M. Torii, M. Sadakane and T. Sano, Bull. Chem. Soc. Jpn., 87, 160 (2014); (g) N. Tsunoji, T. Ikeda, M. Sadakane and T. Sano, J. Mater. Chem. A, 2, 3372 (2014); (h) N. Tsunoji, K. Takahashi, M. Sadakane and T. Sano, Bull. Chem. Soc. Jpn., 87, 1379 (2014); (i) N. Tsunoji, Y. Ide, Y. Yagenji, M. Sadakane and T. Sano, ACS Appl. Mater. Interfaces, 6, 4616 (2014); (j) N. Tsunoji, S. Yuki, Y. Oumi, M. Sekikawa, Y. Sasaki, M. Sadakane and T. Sano, ACS Appl. Mater. Interfaces, DOI: 10.1021/acsami.5b07996.
12) Y. Akiyama, F. Mizukami, Y. Kiyozumi, K. Maeda, H. Izutsu and K. Sakaguchi, Angew. Chem. Int. Ed., 38, 1420, (1999).
13) Z. Li, B. Marler and H. Gies, Chem. Mater., 20, 1896 (2008).
14) Y. Ide, N. Ochi and M. Ogawa, Angew. Chem., Int. Ed. 50, 654 (2011).
15) H. Muraishi and A. Matsuo, Nendo Kagaku 46, 61 (2007).
16) T. Kimura and K. Kuroda, Adv. Funct. Mater., 19, 511 (2009).
17) T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 63, 988 (1990).
18) S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 25, 680 (1993).
19) S. Inagaki, A. Koiwai, N. Suzuki, Y. Fukushima and K. Kuroda, Bull. Chem. Soc. Jpn., 69, 1449 (1996).
20) T. Kimura, T. Kamata, M. Fuziwara, Y. Takano, M. Kaneda, Y. Sakamoto, O. Terasaki, Y. Sugahara and K. Kuroda, Angew. Chem., Int. Ed., 39, 3855 (2000).
21) T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 41, 113 (1996).
22) B. Notari, Stud. Surf. Sci. Catal., 60, 343 (1991).
23) M. Fukuda, N. Tsunoji, Y. Yagenji, Y. Ide, S. Hayakawa, M. Sadakane and T. Sano, J. Mater. Chem. A, 3, 15280 (2015).