スズ化合物は有機合成反応にしばしば利用されている。たとえばR4SnはStilleカップリングとして知られるアルキル化剤として1),またR3SnHは還元剤およびラジカル開始剤として使用される。更に電気陰性度の高いハロゲンと結合したSnX4,SnX2やRnSnX4−nはルイス酸触媒として種々のファインケミカルズの合成反応の促進剤に使われている2)。スズ原子の酸化還元特性を活かすと,スズ化合物は機能性物質としても利用できる。例えば,SnO2は半導体ガスセンサーや透明導電体として,またCO酸化やNOxの還元反応触媒としても働く。
複合酸化物のSnO2–Sb2O4はアリル酸化に,SnO2–CuOはCO酸化に働く。また,SO42−/ZrO2よりも強い超強酸性を示すSO42−/SnO2 (H0=−16.1)は固体酸触媒として,また比較的大きな表面積と貴金属との強い相互作用が期待されて,Pt,Au,Pd,Rhなどの担体として使われる。骨格中にスズ原子を含むゼオライトは,Baeyer–Villiger酸化反応やMeerwein–Ponndorf–Verley還元に有効な触媒として使える。以上のように,スズ化合物は他成分と複合化させることによって,新たな特性を生む可能性を秘めている。ここでは,スズと粘土鉱物モンモリロナイトの複合化の研究成果を紹介する。
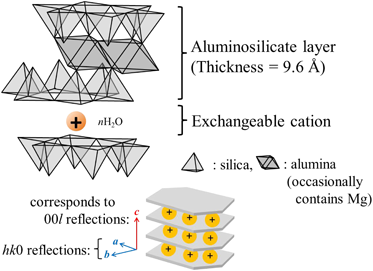
天然粘土鉱物のモンモリロナイト(M-Mont)は,平面アルミナシートを上下から平面シリカシートが挟む基本層を積み重ねた構造をとっている。基本層は負電荷を帯びているため,負電荷を中和するカチオン種(Mn+:天然粘土ではNa+やCa2+)が層間に位置する (Fig. 1)。
この層間のカチオン種はイオン交換が可能であり,特に多価金属イオンで交換したモンモリロナイトは固体酸触媒として利用されている3)。筆者らは1993年にNa-Montと塩化スズ(Ⅳ)水溶液からスズ(Ⅳ)イオン交換操作を施したモンモリロナイト(Sn-Mont)を新たに調製し,従来のアルミニウム(Ⅲ)や鉄(Ⅲ)交換モンモリロナイト(Al-Mont, Fe-Mont)などの多価金属交換モンモリロナイト4)よりも遙かに大きな比表面積・細孔容積をもつユニークな多孔体であり(Table 1),しかも固体酸性も強く,種々の炭素–炭素結合形成反応に優れた酸触媒作用を示すことを明らかにしてきた。
スズ化合物と粘土鉱物がそれぞれもつ特異的な性質が共存した場合に,どのような要因で新たな固体特性が発現するかに興味をもち研究して得られた結果について以下に述べたい。
Table 1. Specific surface areas and pore volumes of the materials.| Material | Specific surface area (m2/g) | Pore volume (mm3/g) |
|---|
| Na-Mont | 20 | 56 |
| Al-Mont | 40 | 74 |
| Fe-Mont | 40 | 75 |
| Ti-Mont | 126 | 130 |
| Sn-Mont | 380 | 270 |
| Sn(OH)4 | 114 | 58 |
| SnO2 | 12 | 46 |
2.1 Sn-Montの構造特異性5)
我々は天然体のNa-MontをSnCl4水溶液に浸してイオン交換した試料を,純水,メタノール水溶液,純メタノールの順で何度も洗浄した後に,乾燥することでSn-Montを調製した。この際洗浄が不十分であると残留する酸成分によってアルミノシリケート層が十分に剥離しないので,丁寧な洗浄操作が大表面積のSn-Montを再現よく得るための秘訣である。
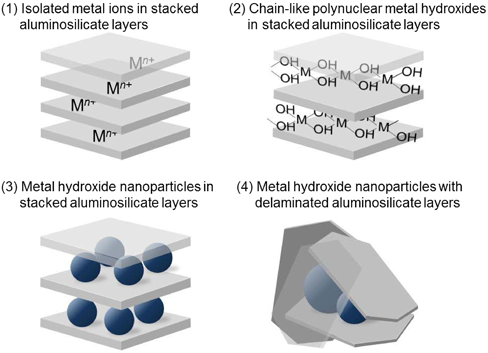
このように調製したSn-Montに含まれるスズ種の形態として,Fig. 2の4種を想定した:(1)孤立したスズ(Ⅳ)イオンがナトリウムイオンに量論的に置き換わって配置する;(2)Ti-Montで提案されている鎖状の酸化チタンのような,鎖状の(水)酸化スズが積層間に形成されている;(3)積層間に(水)酸化スズのナノ微粒子の形で存在する;(4)乱雑に集積したモンモリロナイトのシリケート層に囲まれた(水)酸化スズのナノ微粒子の形で存在する。
スズ(Ⅳ)イオンは水和エンタルピーが大きく,スズイオンに配位した水は脱プロトンしやすい。その結果生じた(水)酸化スズ種はさらに縮合しやすいために,(1)よりも(2)の形態をとりやすいと考えられる。生成した(水)酸化スズどうしの相互作用が,(水)酸化スズと母体のモンモリロナイトシリケート層陰イオンとの相互作用よりも強いならば,さらに縮合度が増して(水)酸化スズ粒子に成長すると,(3)や(4)の形態をとるであろう。
①まずSn-MontをICP分析した結果,含まれるスズの重量は,イオン交換容量から期待される量の14倍も多く含まれていることがわかり,このことからナトリウムイオン4個から1個のスズ(Ⅳ)イオンへの単純なイオン交換ではなく,(1)の可能性がまず否定された。
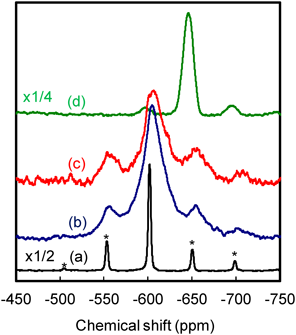
②119Sn-MAS-NMR(Fig. 3)より,Sn-Mont中のスズイオンの化学シフト値は,SnCl4とアンモニア水から調製したSn(OH)4にほぼ一致し,SnCl4とは完全に異なっていた。
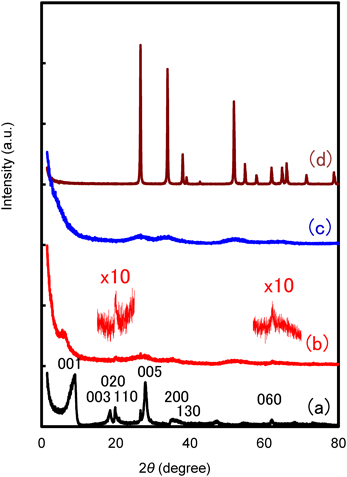
③Sn-Montの粉末X線回折(Fig. 4)からは,Na-Montに見られるようなモンモリロナイトのシリケート層が規則正しく等間隔で積み重なる00lピークは認められなかったが,一枚一枚のシリケート層自身のhk0ピークは残っており,これらの結果からシリケート層が平行ではなく乱れた重なり方をしていると推定された。またシリケート層由来のピークの他に,Sn(OH)4と同様の広幅なピークが観測され,Sn(OH)4の微粒子が形成されていることが示唆された。
④Sn-Montの電子顕微鏡写真(Fig. 5)には,直径3 nm以下の大きさをもつ格子縞が散見された。この粒子径は,粉末X線回折中のブロードな回折ピークから見積もられるSn(OH)4の粒子径にほぼ一致した。
⑤さらにXAFS測定した結果,Sn-MontのXANESおよびEXAFSスペクトルでも,Sn-MontがSn(OH)4の類似構造体を含んでいることが確認された。
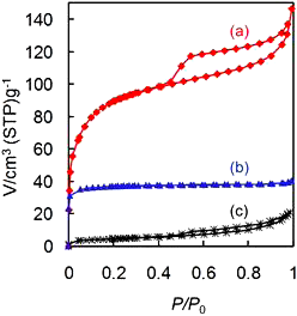
⑥Fig. 6に,Sn-Mont,Sn(OH)4,Na-Montの窒素ガス吸脱着特性を示した。Sn(OH)4はミクロ孔のみをもち,その比表面積は100 m2/gを超えるが,細孔容積は小さい。一方Sn-Montはミクロ孔に加えて多くのメソ孔をもち,比表面積だけでなく細孔容積も大きい。Sn-Montの比表面積は母体のNa-Montの10倍以上大きく,細孔容積もSn(OH)4の4倍程大きい。これらのことより,Sn(OH)4とNa-Montの物理混合物ではないことは明らかである。
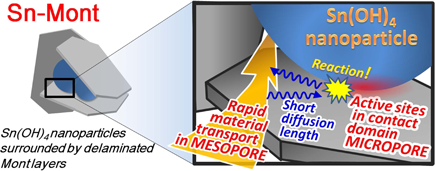
以上の分析結果をまとめると,Sn-Mont中のスズイオンの含有量がイオン交換容量を遙かに超えている点,および窒素ガスの吸脱着等温線の形状(Fig. 6a)から,Sn-Montがとる構造として(1)と(2)の可能性は否定された。更に,粉末X線回折図(Fig. 4)において,イオン交換前のNa-Montがもつ001およびその高次ピークが,スズイオンに交換後失われていることから,(3)よりも(4)の構造をとっていると結論づけた。すなわち,Sn-Montは3 nm以下の水酸化スズのナノ粒子をモンモリロナイト固有の平面シリケート層が乱雑に取り囲む構造と言える。そして水酸化スズのナノ粒子間およびナノ粒子とシリケート層間にミクロ孔とメソ孔が形成されていると考えられ(Fig. 7),大きな比表面積と細孔容積をもつことになる。この構造モデルから考えると,各ミクロ孔はメソ孔に隣接しているので,ミクロ孔領域はそれほど深くないと考えられる。
2.2 Sn-Montの固体酸性
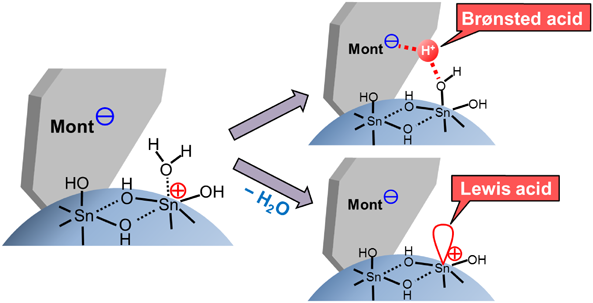
多価金属イオン交換モンモリロナイトは,交換イオンに配位した水分子の解離で生じたプロトンによるBrønsted酸性を示し,解離状態の強弱に応じて酸強度がきまる(Fig. 10)。一方で交換イオンに基づくLewis酸性も持ち合わせる。Sn-Montを固体酸触媒として利用する液相有機合成反応では,微量の水の存在下で反応が進行することが多く,このような反応ではどちらの酸性質が主に機能しているのかを特定するのは一般に難しい。
筆者らは,有機反応速度の大小で粘土鉱物が示す酸触媒能を評価した6)。評価に用いる有機反応としては,反応物に比べて生成物の極性が低く,触媒への生成物の吸着による酸性質の低下を引き起こさない必要性から,アルコール(ROH)とアリルトリメチルシラン(CH2=CHCH2SiMe3)から,アルコールのトリメチルシリルエーテル体(ROSiMe3)とプロピレン(CH2=CHCH3)を生じる反応を選んだ。すなわちこの反応の生成物は両者とも非極性分子であり,生成物による反応阻害は少ないので触媒作用評価に適していると考えられる(Eq. 1)。Eq. 1での酸触媒活性を,反応終了に要した時間で評価した。その結果,Sn-Montは反応時間が最も短く,イオン交換モンモリロナイトの中で最高活性を示すことがわかった(Table 2)。
Table 2. Specific surface areas and pore volumes of the materials.| M-Mont | Specific surface area (m2/g) | Completion time (min) |
|---|
| Sn-Mont | 280 | 10 |
| Fe-Mont | 26 | 35 |
| Al-Mont | 26 | 50 |
| H-Mont | 27 | 70 |
| Cu-Mont | 39 | 100 |
| Zn-Mont | 41 | 360 |
| Ni-Mont | 19 | 380 |
3.1 カルボニル基の活性化とアルドール反応
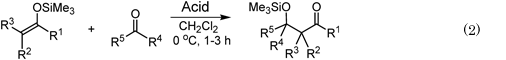
有機合成化学において,有機化合物の炭素骨格をつくりだす炭素–炭素結合が形成される反応はきわめて重要であり,その代表例としてアルドール反応が挙げられる。作曲家と有機化学の教授という二足の草鞋を履いたロシアのボロディンが発見したといわれるこの反応は,強塩基でカルボニル化合物のα位水素を脱プロトンして生じた金属エノラートがアルデヒドに結合し,アルドール付加体を与える。1973年に向山らは不安定な金属エノラートに代えて,単離できるほど安定な珪素エノラートを使う反応形式を提案した(Eq. 2)7)。この形式では珪素エノラートが安定な分,その反応性(求核性)が低いため,相手側のアルデヒドをTiCl4などのルイス酸で活性化する必要がある。これまでにTiCl4の他,種々の均一系ルイス酸触媒(SnCl2,Me3SiOTf,Tf2NHなど)が有効と報告されてきた。
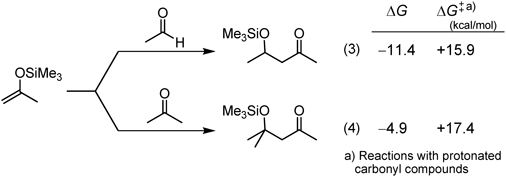
筆者らは,まず1988年に向山アルドール反応に多価金属イオン交換モンモリロナイト(Al-Mont, Fe-Mont)が優れた固体酸触媒として働くことを報告した8)。一般に珪素エノラートは,高い反応性を示すアルデヒドには高収率で付加するが(Eq. 3),嵩高さが増し反応性の低いケトンには付加しにくい(Eq. 4)9)。実際に量子化学計算で見積もると,アルデヒドに比べケトンへの付加反応の方が,より小さな負のΔG値をもち,ΔG≠はより大きな値となり,反応の困難さが伺える。
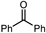
しかし,Sn-Montを固体酸触媒とすると,アルデヒドばかりでなく種々のケトンへのアルドール付加体が高収率で得られることを見出した(Table 3)9)。
Table 3. Mukaiyama-aldol reactions of silicon enolates with ketones.| Silicon enolate | Ketone | Acid catalyst | Yield (%)a) |
|---|
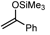 | 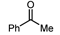 | Sn-Mont | 88 (2) |
| ↑ | ↑ | Ti-Mont | 26 (0) |
| ↑ | ↑ | Al-Mont | 2 (0) |
| ↑ | ↑ | H-Mont | 64 (9) |
| ↑ | ↑ | TMSOTfb) | 0 (45) |
| ↑ | ↑ | BF3·OEt2 | 0 (0) |
| ↑ | 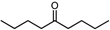 | Sn-Mont | 97 |
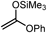 |  | Sn-Mont | 96 |
| a) Figures in parenthesis show yields of the aldol products (β-keto alcohols) .b) Trimethylsilyl triflate |
3.2 カルボニル基へのシアン化物イオンの付加
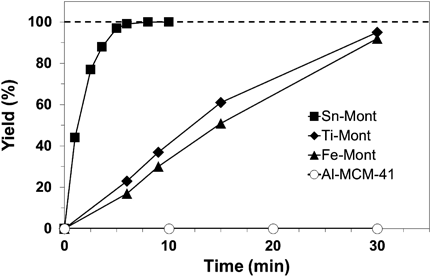
Sn-Montの存在下,Me3SiCN(TMSCN)とアルデヒドまたはケトンを混ぜると,Sn-Montによって活性化されたカルボニル基へのシアン化物イオンの付加反応が促進され,シアノヒドリンのトリメチルシリルエーテル体が生成する(Eq. 5)10)。この生成物はシアノ基を加水分解するとカルボキシル基,還元するとアミノメチル基に変換されるなど,多様な化合物への誘導が可能なので,合成化学上利用価値が高い。酸性を示す種々のモンモリロナイトの使用結果は,アルデヒドに対しては反応速度の大小は見られるもののいずれも高い収率を与えた。一方,よりカルボニル基周りが混み合うために反応性がかなり低下するケトンに対しては,酸触媒能の差が顕在化する。たとえば,低反応性のベンゾフェノンへのTMSCNへの付加反応において,Sn-Montが最高の活性を示した。同じ粘土鉱物のTi-MontやFe-Montに比べ反応速度が大きい(Fig. 8)。また,Sn-Montが層間に含むと考えられるSn(OH)4やSnO2の単独使用では活性が全く出ない10b)。Al-あるいはSn-MCM-41などの固体酸触媒もこの反応には無効であった10b)。
Sn-Montはベンゾフェノン以外の種々の嵩高いケトンへのシアノシリル化反応にも酸触媒としての威力を発揮する(Eq. 5, Table 4)。
Table 4. Cyanosilylation of the following ketones with TMSCN.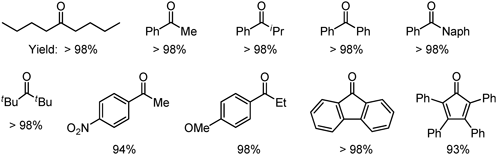 |
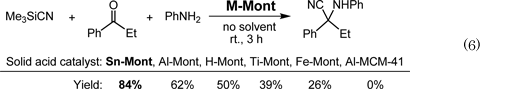
塩基性をもつアミン化合物が共存していても,Sn-Montの酸触媒作用は失われずに,アルデヒド,ケトンから生じるイミンにTMSCNが付加するStrecker反応(Eq. 6)において,他の粘土触媒より優れた触媒特性を示した11)。
3.3 アルコールの活性化とアルキル化剤としての活用
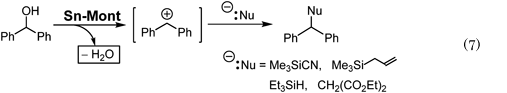
ベンズヒドロール(ジフェニルメタノール,Ph2CHOH)などのベンジル型アルコールの水酸基を酸触媒でプロトン化すると,容易に脱水が起こりジフェニルメチルカチオン(Ph2CH+)を生じやすい。この炭素カチオンは強力なアルキル化剤として働くため,求核剤を加えると両者間で容易に付加反応が進むと期待される。ただしこの場合,脱水で生じた水と求核剤との副反応を抑制する工夫も必要となる(Eq. 7)。
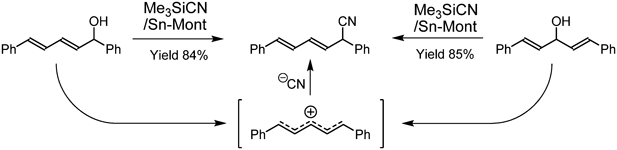
3.2節でも使用した求核性シアン化物イオンとして働くTMSCNの共存下,ベンズヒドロールに触媒量のSn-Montを加えると,シアン化ベンズヒドリル(Ph2CHCN)が生成した(Eq. 8, Table 5)12)。種々の立体的に込み入ったベンジル型アルコールの他,アリル型アルコールにも適用可能であり,特に構造異性体のアリル型アルコールに適用すると,いずれもベンジル位にシアノ基が結合し,生成可能な生成物の中で熱力学的に最も安定なシアン化物体を選択的に与えた(Fig. 9)。このことより,反応はアルコールのプロトン化・脱水反応でカチオン中間体を生じ,熱力学支配でシアノ化反応が進行したと考えられる。
Table 5. Direct cyanation from the following alcohols.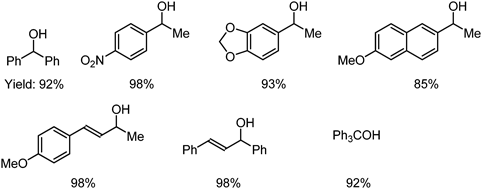 |
同様の反応形式で,求核剤としてアリルトリメチルシラン13),トリエチルシラン(Et3SiH)14),マロン酸エステル15)を使用すると,それぞれ水酸基がアリル基,ヒドリド基,マロニル基で置き換えられた生成物が高収率で得られる。
3.4 Sn-Montの酸触媒作用機構
Sn-Montは,水酸化スズ(Ⅳ)のカチオン性ナノ粒子を,配向を乱しながらモンモリロナイトのアニオン性シリケート層が挟んだ構造をとっている。表面のスズ(Ⅳ)に配位した水がシリケートアニオンと相互作用を受けて解離すればBrønsted酸点となり,その配位水が脱水すればLewis酸点となる(Fig. 10)。
向山アルドール反応への適用例で考えると,これらの酸点にケトンのカルボニル基が配位して活性化され,カチオン性が高まったカルボニル炭素へ珪素エノラートが求核付加し,カルボニル酸素へトリメチルシリル基が転位して,アルドール生成物を与えたと考えられる。モンモリロナイトのシリケート層は求核性が低いマクロアニオンであるため,カチオン性反応中間体の安定化にも大きく寄与し,反応の活性化エネルギーの減少をもたらし,アルドール付加反応速度が高められたと考えられる。同様に,アルコールからSn-Montの酸性作用で生じたカルボカチオンもシリケート層のマクロアニオン効果によって安定化されるので濃度が高まり,SN1型置換反応が効率よく進行したと解釈できる。
従来,イオン交換モンモリロナイトでは,積層した層状シリケートの層間に導入された金属種がその特性に応じた単核イオンあるいは多核イオン形態でインターカレートするのが一般的であった。しかし,Sn-Montでは,水和したスズイオン種が縮合しやすいことによって層間に水酸化スズ(Ⅳ)ナノ粒子を形成し,それをシリケート層が乱雑に取り囲むという特異な形態をとっていることが明らかになった。この特異な構造のために,Sn-Montの表面積や細孔容積は一般の粘土鉱物よりも一桁大きくなり,ミクロ孔・メソ孔をもち,この三次元空洞構造が有機反応の反応場として有効となる。また,シリケート層はマクロアニオンとして,反応場で生成するカチオンの安定化に寄与し,反応の活性化エネルギーの低下をもたらし,種々の触媒反応を促進することに繋がる。
地球の原始から存在するモンモリロナイトは,古くて新しい触媒材料として,多彩な有機合成反応の有効な固体触媒として,利用法がますます広がるであろう。
引用文献References
1) M. Kosugi, K. Sasazawa, Y. Shimizu, T. Migita, Chem. Lett., 301 (1977), D. Milstein, J. K. Stille, J. Am. Chem. Soc., 100, 3636 (1978).
2) H. Yamamoto, “Lewis Acids in Organic Synthesis”; Wiley-VCH: Weinheim, Germany, 2000.
3) 触媒便覧/触媒学会編,講談社サイエンティフィク,p. 382 (2008).
4) Y. Izumi, M. Onaka, Adv. Catal., 38, 245 (1992).
5) Y. Masui, J. Wang, K. Teramura, T. Kogure, T. Tanaka, M. Onaka, Micropor. Mesopor. Mater., 198, 129 (2014).
6) M. Onaka, Y. Hosokawa, K. Higuchi, Y. Izumi, Tetrahedron Lett., 34, 1171 (1993).
7) T. Mukaiyama, K. Narasaka, K. Banno, Chem. Lett., 2, 1011 (1973); T. Mukaiyama, K. Banno, K. Narasaka, J. Am. Chem. Soc., 96, 7503 (1974).
8) M. Kawai, M. Onaka, Y. Izumi, Bull. Chem. Soc. Jpn., 61, 1237 (1988).
9) S. Takehira, Y. Masui, M. Onaka, Chem. Lett., 43, 498 (2014).
10) (a) K. Higuchi, M. Onaka, Y. Izumi, Bull. Chem. Soc. Jpn., 66, 2016 (1993); (b) J. Wang, Y. Masui, K. Watanabe, M. Onaka, Adv. Synth. Catal., 351, 553 (2009); (c) J. Wang, Y. Masui, T. Hattori, M. Onaka, Tetrahedron Lett., 53, 1978 (2012).
11) J. Wang, Y. Masui, M. Onaka, Eur. J. Org. Chem., 1763 (2010).
12) J. Wang, Y. Masui, M. Onaka, ACS Catal., 1, 446 (2011).
13) J. Wang, Y. Masui, M. Onaka, Tetrahedron Lett., 51, 3300 (2010).
14) M. A. Tandiary, Y. Masui, M. Onaka, Tetrahedron Lett., 55, 4160 (2014).
15) J. Wang, Y. Masui, M. Onaka, Synlett., 2493 (2012).