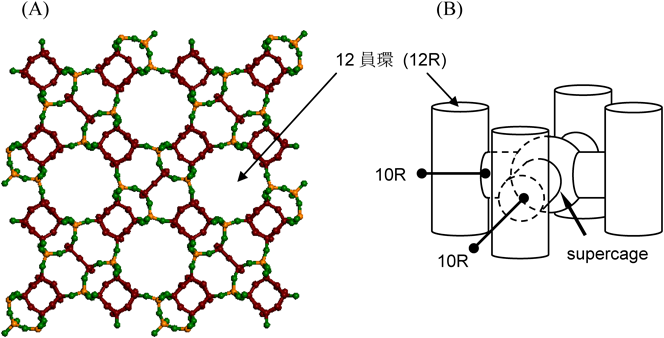
5. YNU-2Pから規則性ミクロ多孔体YNU-2への変換
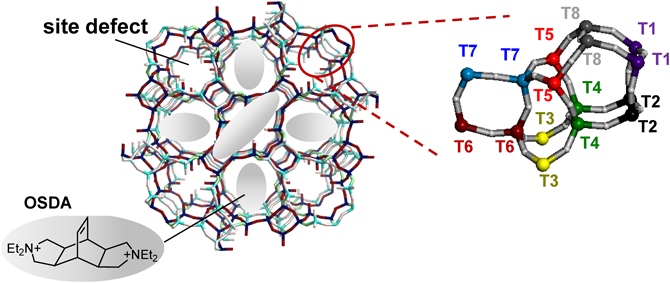
YNU-2Pは得られたものの,有機物を常法で除いただけで規則性多孔体YNU-2を直接得ることは出来なかった。すなわち,400°C以上で焼成すると結晶構造が崩壊した20,26)。高分解能粉末X線回折データに基づく精密構造解析20)によれば,YNU-2PではMSEユニットセル内の8種類のTサイト(図2)のうちT6, T7サイトの充足率gがいずれも0.40と特に低く,T1, T3でそれぞれ0.84, 0.90, それら以外(T2, T4, T5, T8)では1.0であった。そして,ユニットセル(Tサイト総数112)あたり約13個の欠陥が存在し,OSDAが図2に示すように包接されて骨格を支えていた(産総研・池田拓史博士による)。10%を超える欠陥率であり,YNU-2Pの骨格構造が焼成により崩壊するのは,この欠陥の多さに起因する。
そこで,ポスト処理によりその欠陥サイトの修復を試みた。具体的には,1.0 mol L–1硝酸およびSi(OEt)4の混合物が入ったオートクレーブにYNU-2Pを懸濁させ,160°Cで24時間加熱した。その結果,ポスト処理後の固体試料は450°Cで焼成しても高い結晶性を示すMSE構造を保持するようになった。こうして,新しい規則性多孔体YNU-2が得られた。YNU-2Pに存在していた欠陥サイトには確かにSiが挿入されていた。したがって,ポスト処理による欠陥修復を経て熱的に安定なゼオライト骨格が形成されたと考えられる20,26)。
この時点で,Si源ではなく各種のヘテロ元素源を用いることにより,アトムプランティングが可能ではないか,欠陥の多さを逆手にとってYNU-2Pが各種アトムプランティングの土台になり得るのではないかという着想を得たため,手始めにAlの導入を試みた。Si(OEt)4の代わりにAl(OEt)3を用いた類似の処理を行うと,やはり骨格の安定化が起こったものの,Alの導入量は極めて少なかった21)。つまり,骨格の安定化に寄与しているのは主としてSiであり,ここで,骨格Siのマイグレーションの可能性が浮上した。この骨格Siのマイグレーションを検証するために,系外からSi源もヘテロ元素源も添加せず,YNU-2Pを1.0 mol L−1硝酸中で加熱処理したところ,やはり焼成後にYNU-2が得られた。Si種のマイグレーション自体は比較的古くから知られている現象ではあるが,新規物質においてもこの現象を目の当たりにすることとなった。結局,YNU-2Pはスチーミング処理だけでも骨格が安定化することがわかり,29Si MAS NMRにおけるQ3ピークの減少などから,YNU-2PのSi種のマイグレーションが起きていることが明らかとなった26)。
6. YNU-2へのTiの導入によるTi-YNU-2触媒の創製29)
YNU-2骨格へTiを導入する方法は複数考えられるが,我々はまず穏和な(ゆるい)スチーミング条件で必要最小限のSiマイグレーションを起こして骨格を安定化し,残存するできるだけ多くの欠陥にTiを導入するという方針をたてた。Ti導入には,TiCl4を気相から供給する手法を用いることとした15,17)。これは,並行して進めているTi-MCM-68の研究開発でも多用している方法である31)。
6.1 スチーミング条件の検討
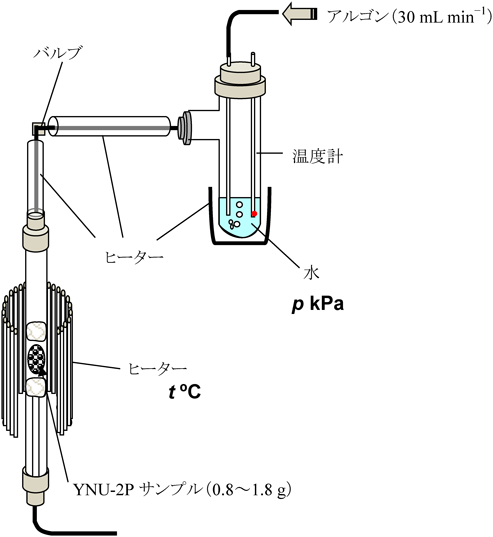
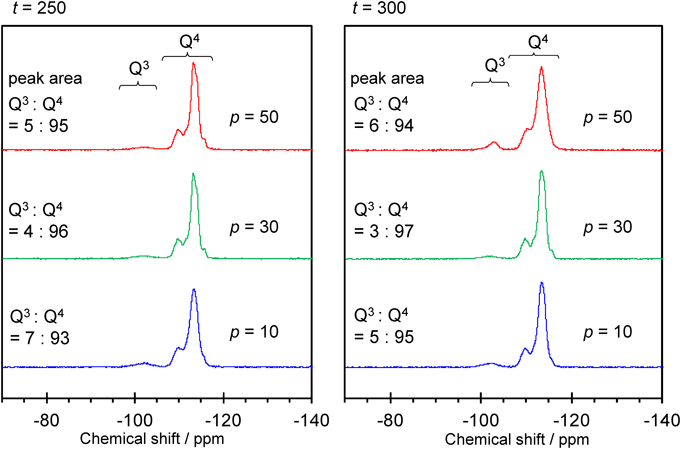
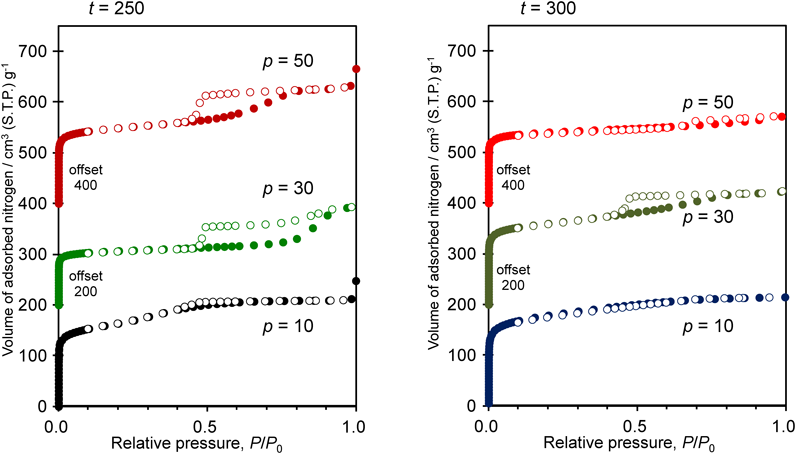
Ti導入の前に,YNU-2Pに対するスチーミング条件を詳しく検討した。図3に示すスチーミング装置を用い,水蒸気を異なる温度(t°C; t=200, 300)および分圧(p kPa; p=10, 30, 50)で24時間流通させた。これらの条件下でのスチーミングはいずれも有効で,処理後のサンプルは,包接されている残存有機物を焼成(450°C, 3 h)によってすべて除いても骨格を維持した。これに対し,200°C以下のスチーミングでは安定化効果がみられなかった。水蒸気分圧が10 kPaのとき,スチーミング直後の有機物含有率(熱重量分析による)は,処理温度200, 250, 350°Cにおいてそれぞれ21.1, 17.1, 12.2%であり,元々のYNU-2P中のOSDA含有率が23.0%であったことを考えあわせると,有機物がある程度除去されるような温度ではじめてSi種のマイグレーションによる骨格安定化が可能になると言える。ここで,スチーミングによる骨格安定化の後に焼成(450°C, 3 h)で有機物を完全に除いて最終的に得られた多孔体をYNU-2 (t, p)と定義する(括弧内のt, pは上で述べたスチーミング温度と分圧である)。各YNU-2 (t, p)の焼成後の29Si MAS NMRを図4に示す。メジャーなQ4ピークとマイナーなQ3ピークの存在が,骨格の安定化がなされたことと共に依然として欠陥が残存していることを示している。スチーミング条件を厳しくするにつれ,欠陥に由来するQ3ピークの割合が減少し,Q4ピークがシャープになっている。このことから,スチーミングを厳しく行うことでSi種のマイグレーションが活発化し,欠陥のより少ない安定な骨格構造が得られることが示唆される。また,スチーミング条件によっては窒素吸脱着等温線上でヒステリシスループが観測されるようになった(図5)。ヒステリシスループは直径約4 nm以上のメソ孔が存在する場合に観測されるものであり,YNU-2 (250, 50), YNU-2 (250, 30), YNU-2 (300, 30)の各サンプルにおいて条件に合うメソ孔の生成が示唆された。さらに,このことはTEM観察によっても裏付けられた。
原子・分子レベルで見た場合,Si種のマイグレーションは基本的にはSi-O結合の加水分解から始まる。この際,欠陥に隣接するSiが,水分子との親和性と立体障害のいずれの観点からも,加水分解を受けやすいと考えるのが妥当である。骨格との共有結合がすべて失われたSi(OH)4種が移動すると考えるのが最も自然であるが,このような化学種が移動できるようなスペースもまた必要と考えられ,先に述べた有機物除去の必要性は,このスペースの提供とも関係がある。つまり,有機物が除かれれば,ミクロ孔を通じてSi種は移動できると考えられる。
より巨視的な観点からは,欠陥の動きはSi種の動きとは逆方向で,粒子内のいろいろな場所から粒子の末端へ向かって集約されていくと考えることができる。そして粒子末端が粒界であった場合にメソ孔として観測されるようになるのではないかと推測される。適度なスチーミング条件下でメソ孔が生成・成長していき,その過程の一時期に吸脱着測定におけるヒステリシスループが観測されるのかもしれない。
再び微視的観点にもどると,バルクでの空隙があったとしても骨格安定化がなされており,XRDもシャープなので,原子配列の規則性は高いといえる。スチーミング条件の変化は,Si種のマイグレーションの度合いを変化させ,後にTiが導入されるサイトの位置や分布,あるいは親疎水性などのマクロ物性に影響し得る。後述するように,これがひいては触媒性能に影響することになる。
6.2 骨格へのTi導入とフェノールの過酸化水素酸化対する触媒性能
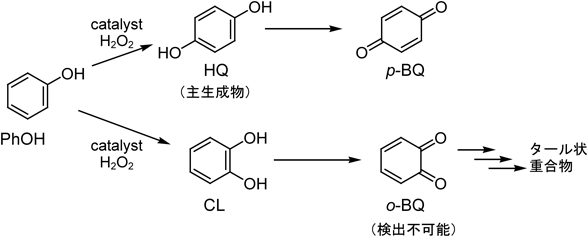
先に得られたYNU-2 (t, p)に対し,500°Cで加熱しながらTiCl4/Arを1時間流通させてTi-YNU-2 (t, p)触媒を調製した。次いで,冒頭で述べたH2O2を酸化剤として用い,フェノールの酸化反応に対する触媒性能を検討した。結果を表1に示す。フェノールの酸化はスキーム2に示したように,併発および逐次反応から成っている。一段目の水酸化は併発反応であり,ヒドロキノン(HQ)とカテコール(CL)の間の相互転換(異性化)は起こらないことを別途確かめた。また,レゾルシノールの生成も全く見られなかった。さらに,逐次酸化生成物のうちオルトベンゾキノン(o-BQ)は不安定なため,仮に生成しても容易に重合物となって検出できないことが自明であり,実際にも検出されなかった。表1に示すとおり,スチーミング温度は300°Cよりも250°Cの方が好ましく,水蒸気分圧は低めの10 kPaが検討した中では最適であった。結果として,Ti-YNU-2 (250, 10)がTON 923, パラ選択率92%と,非常に高い活性とパラ選択性を示した。
表1 種々のチタノシリケート触媒を用いたフェノールの過酸化水素酸化a| entry | catalystb | Ti-contentc/mmol (g-cat.)−1 | TONd | yielde(%) | para-sel.f(%) | H2O2 |
|---|
| total | HQ | CL | p-BQ | conv. (%) | eff. g(%) |
|---|
| 1 | Ti-YNU-2 (250, 10) | 0.18 | 923 | 74.3 | 58.4 | 6.1 | 9.8 | 92 | 88.4 | 83.2 |
| 2 | Ti-YNU-2 (250, 30) | 0.16 | 786 | 59.2 | 45.2 | 7.4 | 6.7 | 88 | 82.9 | 71.5 |
| 3 | Ti-YNU-2 (250, 50) | 0.16 | 704 | 53.0 | 38.4 | 6.5 | 8.1 | 88 | 90.3 | 58.7 |
| 4 | Ti-YNU-2 (300, 10) | 0.20 | 444 | 42.2 | 27.5 | 4.6 | 10.1 | 89 | 67.8 | 62.2 |
| 5 | Ti-YNU-2 (300, 30) | 0.10 | 127 | 5.7 | 2.6 | 1.4 | 1.7 | 76 | 20.1 | 28.2 |
| 6 | Ti-YNU-2 (300, 50) | 0.07 | 118 | 4.1 | 1.5 | 1.0 | 1.7 | 76 | 14.2 | 29.1 |
| 7 | Ti-MCM-68-cal | 0.25 | 272 | 32.3 | 22.2 | 8.3 | 1.8 | 74 | 63.3 | 51.1 |
| 8 | Ti-MCM-68 | 0.25 | 150 | 17.1 | 11.1 | 5.5 | 0.5 | 58 | 34.0 | 50.1 |
| 9 | TS-1 | 0.36 | 50 | 8.4 | 4.7 | 3.7 | 0.0 | 56 | 25.2 | 33.4 |
a Reaction conditions: phenol (PhOH), 21.05–21.85 mmol; catalyst, 20 mg; H2O2, 4.12–4.55 mmol; temperature, 100°C; time, 10 min.
b First and second values in parentheses are steaming temperature, t/°C and steam pressure, p/kPa, respectively.
c Determined by ICP analysis.
d Turnover number (moles of [hydroquinone (HQ) + catechol (CL) + p-benzoquinone (p-BQ)] per mole of Ti site).
e Product yields based on added H2O2 after exhaustive acetylation of the products with excess (CH3CO)2O-K2CO3, the derivatized products were analyzed by GC (0.25 mm×30 m×1.00 µm DB-1 column, internal standard: anisole, detector: FID).
f Selectivity to para-isomers of dihydroxybenzenes and quinones (moles of [HQ+p-BQ] per moles of [HQ+CL+p-BQ]).
g Efficiency of H2O2 utilization (moles of [HQ + CL + p-BQ] per mole of H2O2 converted). |
比較のために従来型のTi-MCM-68およびTS-1の結果も表1に記載した。Ti-MCM-68は熱処理してTi-MCM-68-calとすることで,活性と選択性が向上したが,Ti-YNU-2には及ばなかった。また,TS-1は有効ではあるものの,この条件ではTi-MCM-68よりも低活性であり,10員環細孔しかもたないことに起因して選択性も低かった。
6.3 高い活性発現の原因
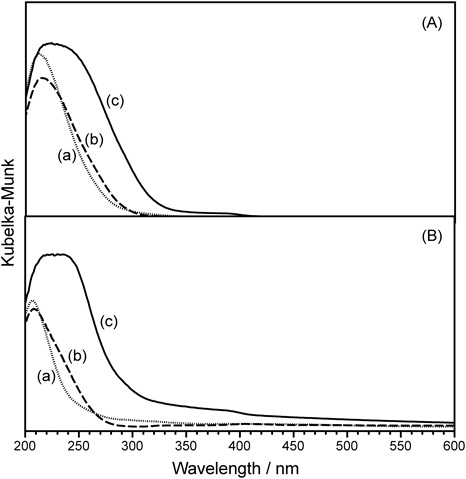
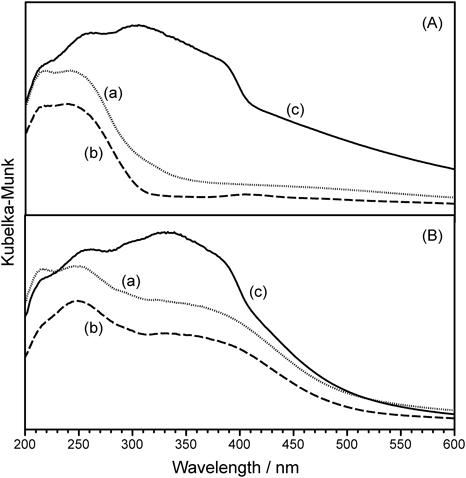
Ti-YNU-2の非常に高い活性の原因をつきとめるため,活性点構造の様子を探った。TS-1, Ti-MCM-68, Ti-YNU-2それぞれの拡散反射紫外可視(DR UV-vis)スペクトルを,擬in situセル35)を用いて測定し,図6に示す結果を得た。図6Aが真空排気前,図6Bが400°Cでの真空排気後のスペクトルである。まず図6Aでは,全てのチタノシリケートで210 nm付近に4配位Ti種(closedおよびopenサイト)由来のピークが観測された。しかし,Ti-YNU-2の場合のみ,250~290 nm付近にも肩が観測された。この領域のピークは5配位および6配位Ti種に帰属される4)ため,Ti種周りの配位状態はTS-1やTi-MCM-68と比較して最初から大きく異なることがわかった。次に400°C, 2 h真空排気した後のスペクトル(図6B)をみると,TS-1およびTi-MCM-68の210 nm付近のピークは吸着水除去の影響でよりシャープになり,典型的な4配位Ti種のパターンとなっていた。これに対してTi-YNU-2の場合はこれらの他に,250~290 nm付近のピークがややすそ野が狭まったものの依然として顕著であった。TS-1に関する既往の報告4)と照らし合わせると,5配位および6配位Ti種とともに,Tiにヒドロキシ基が一つ以上結合した4配位Ti種が比較的多く存在していることが示唆された。
次に,真空排気後(図6B)のサンプルを擬in situセルに入れたまま水を少量加え,再びDR UV-vis測定を行い,図7Aに示すスペクトルを得た。こうして得たTS-1とTi-MCM-68のスペクトル(図7A-a, b)は,図6B-cと非常によく似ており,5配位および6配位Ti種またはヒドロキシ基を一つ以上持つ4配位Ti種(openサイト)が増加したことを示していた。これに対し,Ti-YNU-2の場合は全く異なるスペクトルが得られた。波長300~400 nmに現れたブロードで大きなピークは,ヒドロキシ基を一つ以上持つ5配位および6配位Ti種と帰属された。そこで次に,擬in situセル中,真空排気後のサンプルに対して水の代わりに31% H2O2水溶液を少量加え,再びDR UV-vis測定を行った。すると,Ti-YNU-2の場合に特に大きいものの,結局どのサンプルに対しても300~400 nmの範囲にブロードなピークが現れた(図7B)。これは,チタノシリケート触媒の作動状態におけるTi-OOH種によるものと考えられる。Ti-YNU-2は水を加えただけでこれに近い状態であったことを考えあわせると,Ti-YNU-2は元々この状態になりやすい傾向を有しており,活性化状態への「準備が整った」触媒であると言える。これが,Ti-YNU-2が非常に高い活性を示す理由の一つであると考えている29)。
なお,Ti-YNU-2におけるTiサイトの配位数については,XAFSによっても上記の解釈を裏付ける結果が得られている(未発表データ)。
6.4 高いパラ選択性発現の原因
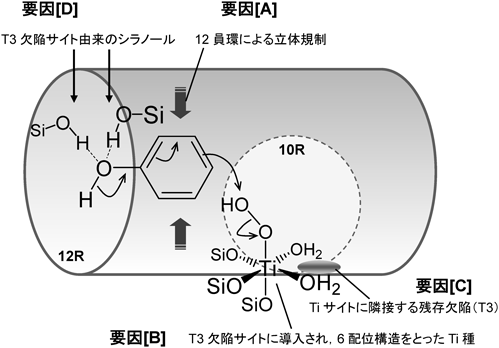
ゼオライト細孔によるパラ選択性は基本的には細孔の立体規制により発現する(図8の要因[A])。しかし,Ti-YNU-2の非常に高いパラ選択性の発現要因はそれだけでは説明し難い。そこで,次のような仮説を提唱する29)。断片的な結果をつなぎ合わせ,骨格模型(図9)を活用して行った推測であるが,各原子間距離と相互作用・電子の動き・結合の開裂と形成は化学的に妥当な範囲を逸脱してはいない。今後,計算機化学の支援を得れば,より緻密な推測が可能になると期待される。
5節で述べたように,YNU-2Pは図2のT6とT720,26)(ref. 22および23ではそれぞれT7とT8)にかなりの欠陥を有している。つまり,T6とT7の位置にあるべきSiが抜けている。(以下の説明の視覚化は紙面では限界があるが,図9の骨格模型を活用することにより状況把握が容易になる。)これらT6, T7サイトは12員環ストレートチャンネルに面していないが,隣接するT3とT5からそれぞれSiが供給された場合,12員環に面するT3, T5に欠陥が移る。今,簡単のためにT3に欠陥ができるとする。そうすると,Ti導入によって12員環に活性点が生じることになる。また,二つのT3が互いに隣接しているので,連続した欠陥が生じ得る。そのうち一つのT3のみにTiが挿入されれば,ヒドロキシ基をもつ(あるいはH2Oが配位した)骨格内Tiとなり,このようなTiは骨格からはずれることなく配位数や立体構造の変化(つまりテトラヘドラルな4配位からオクタヘドラルな6配位構造への変化;図8の要因[B])が容易になるとともに,反応剤や基質が接近するスペースも確保できる要因[C])。もしこうした環境にあるTiサイト,すなわちシラノールネストに近接するTiサイトがTS-1骨格内にも存在した場合,基質の配位による歪んだ6配位Tiの形成につながることが,DFT計算により示唆されている36)。ここまでは,前節で述べた高い活性発現の要因とも言える。
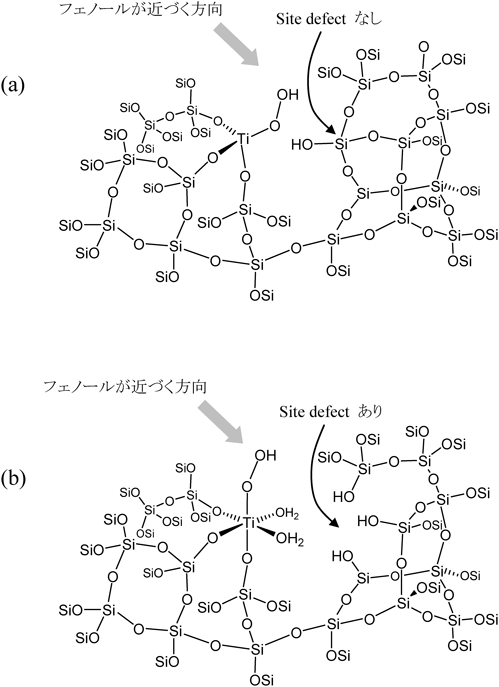
これらを踏まえ,一部の重複も含めて図8の説明をさらに進めると次のようになる。例えばT3欠陥サイトだけをとっても,細孔内の規則的な位置に複数存在するため,それらが「活性点挿入の場(要因[B])」,「反応剤が活性点に近づくためのスペース(要因[C])」,「基質を(水素結合などによって)捕捉する官能基(要因[D])」などとしての役割を適宜果たしていると考えると,優れた触媒性能を発現する原因として一つの説明が成り立つ(T5についても同様の可能性があるが,省略する)。すなわち,(1)第一のT3欠陥サイトにTiが挿入され,その隣にもT3欠陥サイトが残存する(要因[C])のでTiサイトは構造に自由度をもつ(要因[B]),(2)第二,第三のT3欠陥サイト(シラノール)が水素結合を介してフェノール分子を捕捉し,弱い相互作用でフェノールの向きと位置を制限する(要因[D]),(3)フェノール水酸基のパラ位がTi-OOH種に特に接近するため,パラ置換の反応速度が特に増大する。なお,(1)の理解を助けるために,過酸化水素で活性化されたTi-YNU-2の部分構造(隣にsite defectが残存するTiサイト)を概念的に示し,従来型チタノシリケート(Ti-MCM-68, TS-1など)のいわゆるopen site4)の場合と比較したのが図10である。
以上(1)~(3)は,これまで「細孔による立体規制」一辺倒であった「形状選択性」の発現機構37)に一石を投じる考え方である。酵素反応において酵素が基質を分子認識して捕捉する事により,ランダムな分子運動を制限してエントロピーを減少させ,基質同士,基質と反応剤もしくは基質と酵素の反応部位を近づけることで反応の活性化エネルギーを大きく低下させる,いわゆるエントロピー・トラップに似ているが,単なる酵素の模倣ではなく,ゼオライト本来の特質である「ミクロ孔による立体規制(図8の要因[A])」が掛け合わさっている点で,ユニークな系と言える。今後の検証と拡張を望みたい。
なお,Ti-YNU-2触媒は反応後に水洗および焼成(550°C, 4 h)を経て再使用が可能であり,再使用中にもTiのリーチングが認められなかったことから,実用的にも有望である29)。
引用文献References
1) B. Notari, Adv. Catal., 41, 253–334 (1996).
2) T. Tatsumi, Curr. Opin. Solid State Mater. Sci., 2, 76–83 (1997).
3) I. W. C. E. Arends, R. A. Sheldon, Appl. Catal., A: General, 212, 175–187 (2001).
4) P. Ratnasamy, D. Srinivas, H. Knoezinger, Adv. Catal., 48, 1–169 (2004).
5) P. Wu, T. Tatsumi, Catal. Surv. Asia, 8, 137–148 (2004).
6) R. Noyori, M. Aoki, K. Sato, Chem. Commun., 1977–1986 (2003).
7) M. Taramasso, G. Perego, B. Notari, US Patent, 4410501 (1983).
8) J. S. Reddy, R. Kumar, P. Ratnasamy, Appl. Catal., 58, L1–L3 (1990).
9) M. A. Camblor, A. Corma, A. Martínez, J. Pérez-Pariente, J. Chem. Soc., Chem. Commun., 589–590 (1992).
10) J. C. van der Waal, P. J. Kooyman, J. C. Jansen, H. van Bekkum, Microporous Mesoporous Mater., 25, 43–57 (1998).
11) T. Tatsumi, N. Jappar, J. Phys. Chem., 102, 7126–7131 (1998).
12) A. Tuel, Zeolites, 15, 236–242 (1995).
13) D. P. Serrano, H.-X. Li, M. E. Davis, J. Chem. Soc., Chem. Commun., 745–747 (1992).
14) P. Wu, T. Komatsu, T. Yashima, J. Phys. Chem., 100, 10316–10322 (1996).
15) C. B. Dartt, M. E. Davis, Appl. Catal., A: General, 143, 53–73 (1996).
16) M.-J. Díaz-Cabañas, L. A. Villaescusa, M. A. Camblor, Chem. Commun., 761–762 (2000).
17) P. Wu, T. Tatsumi, T. Komatsu, T. Yashima, J. Phys. Chem. B, 105, 2897–2905 (2001).
18) P. Wu, T. Tatsumi, Chem. Commun., 1026–1027 (2002).
19) P. Wu, T. Miyaji, Y. Liu, M. He, T. Tatsumi, Catal. Today, 99, 233–240 (2005).
20) Y. Koyama, T. Ikeda, T. Tatsumi, Y. Kubota, Angew. Chem. Int. Ed., 47, 1042–1046 (2008).
21) 窪田好浩,稲垣怜史,触媒,51, 304–309 (2009).
22) Ch. Baerlocher, L. B. McCusker, D. H. Olson, Atlas of Zeolite Framework Types, 6th Ed., Elsevier, Amsterdam, 2007; http://www.iza-structure.org/databases/
23) D. L. Dorset, S. C. Weston, S. S. Dhingra, J. Phys. Chem. B, 110, 2045–2050 (2006).
24) D. C. Calabro, J. C. Cheng, R. A. Crane, Jr., C. T. Kresge, S. S. Dhingra, M. A. Steckel, D. L. Stern, S. C. Weston, US Patent, 6049018 (2000).
25) J. G. Moscoso, D.-Y. Jan, US Patent, 7922997 (2011).
26) T. Ikeda, S. Inagaki, T. Hanaoka, Y. Kubota, J. Phys. Chem. C, 114, 19641–19648 (2010).
27) S. Inagaki, Y. Tsuboi, Y. Nishita, T. Syahylah, T. Wakihara, Y. Kubota, Chem. Eur. J., 19, 7780–7786 (2013).
28) Y. Kubota, K. Itabashi, S. Inagaki, Y. Nishita, R. Komatsu, Y. Tsuboi, S. Shinoda, T. Okubo, Chem. Mater., 26, 1250–1259 (2014).
29) M. Sasaki, Y. Sato, Y. Tsuboi, S. Inagaki, Y. Kubota, ACS Catal., 4, 2653–2657 (2014).
30) T. Shibata, S. Suzuki, K. Komura, Y. Kubota, Y. Sugi, Kim, S. Gon, Microporous Mesoporous Mater., 116, 216–226 (2008).
31) Y. Kubota, Y. Koyama, T. Yamada, S. Inagaki, T. Tatsumi, Chem. Commun., 44, 6224–6226 (2008).
32) S. Inagaki, K. Takechi, Y. Kubota, Chem. Commun., 46, 2662–2664 (2010).
33) S. Park, Y. Watanabe, Y. Nishita, T. Fukuoka, S. Inagaki, Y. Kubota, J. Catal., 319, 265–273 (2014).
34) M. Matsukata, M. Ogura, T. Osaki, P. R. H. P. Rao, M. Nomura, E. Kikuchi, Topics in Catalysis, 9, 77–92 (1999).
35) A. Zecchina, S. Bordiga, C. Lamberti, G. Ricchiardi, C. Lamberti, G. Ricchiardi, D. Scarano, G. Petrini, G. Leofanti, M. Mantegazza, Catal. Today, 32, 97–106 (1996).
36) D. H. Wells, Jr., W. N. Delgass, K. T. Thomson, J. Am. Chem. Soc., 126, 2956–2962 (2004).
37) S. M. Csicsery, Pure Appl. Chem., 58, 841–856 (1986).
38) M. M. J. Treacy, Micropor. Mesopor. Mater., 58, 1–2 (2003).