近年,元素戦略や新奇触媒材料開発などの観点から,微粒子金属を原子レベルで混合することによって創製された微粒子合金による,新奇な電子状態の創出をめざした“化学”が注目されている1,2)。元素に新奇な電子状態を賦与する方法は合金化だけではない。アメリカのCastlemanのグループは電子の足し算によって,もとの元素とは違う元素の特性を作り出すことができることを報告している。たとえば,Zr(電子数4個:5s2+4d2)と酸素(6個:2s2+2p4)の組み合わせでPd(10個:5s2+4d8)の特性を作り出せるとのことである。彼らは,これらを,super-ionおよびsuper-atomと名付けている3,4)。上述した,合金でも類似した現象が発現する。これらは,いずれも,21世紀の錬金術と言っても良いと考える。我々は,これまで,ゼオライト場によって創出される金属イオンの新奇な電子状態の創製(金属イオンによる新物質デザイン)を目指して研究を行ってきた。これらの現象を研究することによって,ゼオライト触媒上で起こる触媒作用の本質を解明することができると期待している。ゼオライトを新奇な反応場として用いた興味深い現象として,たとえば,岩本らによる先駆的な研究,CuMFIによるNOの分解反応などがある5)。実際,サブナノサイズを有する空間(サブナノ空間と略称)中にイオン交換された一部の金属イオンはバルクのそれとは全く異なる性質を示すのである。このような特異な状態をとる金属イオンがNO分解の活性点として機能していると考えることができる。我々は,ゼオライト中にイオン交換された金属イオンの状態は不均一であるように理解していたが,東ソー製のモルデナイト(MOR: Si/Al=10)やMFI(Si/Al=11.9)試料を銅イオンでイオン交換し,その後,873 Kで真空引した試料においては,CO分子に対する吸着熱の測定から,エネルギー的に均一なサイトが少なくとも二種類存在することを見いだしたことから,ゼオライト中の銅イオンの特性解析が可能と判断し,この系に注目して研究を行ってきた6,7)。そして,CuMFI中の吸着サイトはCu+であり,このCu+種は,室温で,窒素分子やXeに対しても驚くべき特性を示すことも明らかにした8–13)。もちろん,たとえば,バルクのCu2Oはこれらの特性を全く示さない。このことは,ゼオライト空間を利用することで,全く新しい特性を示すCu+種を作り出したことになる。
我々は,このような固体表面上にイオン交換された金属イオンあるいはこのような金属イオンと特異的に相互作用することによって形成された種を新奇な化合物という視点で捉えている。このサブナノ空間中のイオンが,触媒の活性点として機能していると理解している。これらの現象について,よく言われる理解としては,サブナノ空間内で作り出された①配位不飽和な金属イオンの状態とか②金属イオン周りの配位歪みの効果であるというとらえ方をされる。しかし,注目すべき点は,ゼオライト中の金属イオンの特徴の電子的な研究はほとんど理解されていないことである。
このような考え方を基盤として,我々は,ゼオライト中の二価イオンの状態解明を目指し,還元に対して比較的不活性であると考えられる二価亜鉛イオンを利用して水素やメタンの活性化現象14)を検討した。その結果,ZnMFI上で水素やメタンの活性化が室温付近で起こること,その活性サイトはゼオライト中の特異なAlの原子配列をもつサイトであるという結論を得た。ここでは,これらの興味深い現象について解説したい。
2. ZnMFI上で観測される室温付近での水素の活性化15–18)
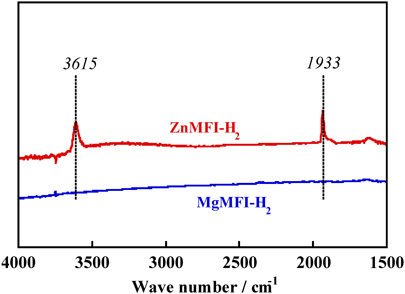
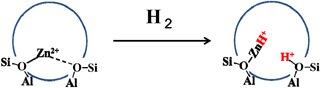
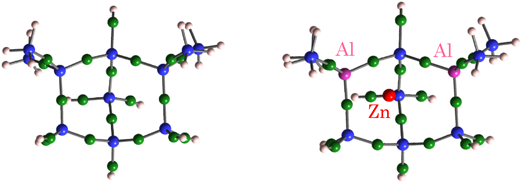
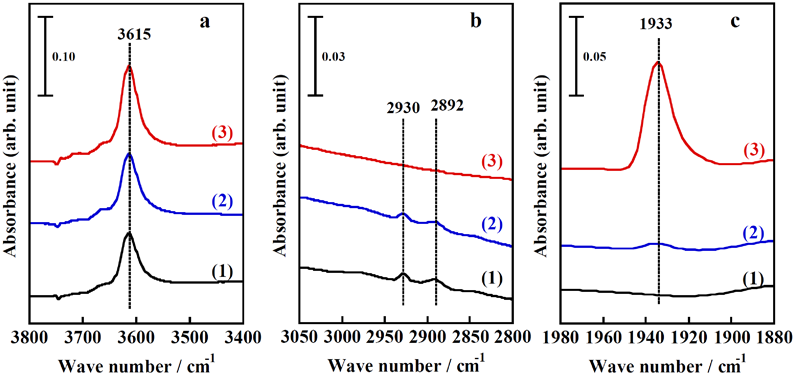
亜鉛イオン交換MFI型ゼオライト試料(ZnMFI-95と略記:なお,数字の「95」はイオン交換量(%)を表す)を873 Kで真空排気した試料は,300 Kでさえ,水素の解離(不均一結合開裂)を引き起こす。すなわち,次式に従って,ZnMFI上のZn2+は水素分子を活性化し,–ZnH (1933 cm−1)と–OH (3615 cm−1)種を形成する(図1)。その際,使用した試料のイオン交換量が95%であること,XANES(X-ray absorption near-edge structure)スペクトルのデータは二価の亜鉛種がイオン交換されていることを示唆することなどを考慮した。
(ここで,ZAとZBはAl原子を含むゼオライト骨格を意味する。)
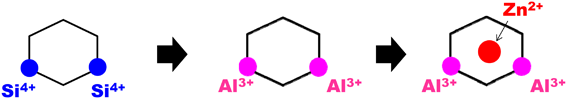
ZnMFI-H2系の反応をもう少し理解し易いモデル図として描くとスキーム1のようになる。すなわち,ZnMFl-95においてゼオライト中の二つのSi原子を二つのAl原子で置換することによって生じた二個の負電荷を一個の二価亜鉛イオンで同時に電荷補償を行うと考える。このサイト上で水素分子が不均一開裂され,–ZnHと–OH種が形成される。
また,反応性を比較するために,金属イオンとしてZn2+イオンと比較的類似した性質を有するMg2+イオンでイオン交換したMgMFI-82試料について,同様の実験を行い,水素の活性化について比較した。その結果を図1に示す。これより,MgMFI系では水素の活性化は全く生じないことは明らかである。
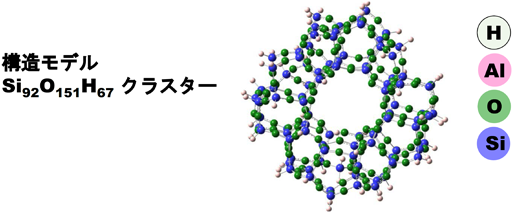
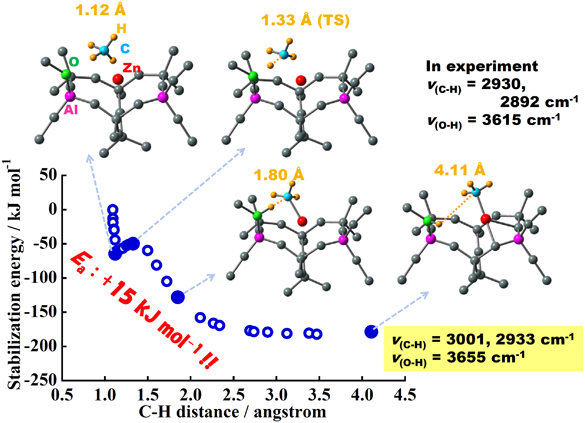
次に,ZnMFI中の水素活性化サイトの状態および活性化機構に関する詳細な知見を得ることを目的としてDFT計算による解析を行った。上述したように,MFI中にイオン交換された単核のZn2+が活性点として機能することがわかったので,MFI中にイオン交換されたZn2+の状態を計算で再現し,その種による水素・メタン活性化能を評価した。イオン交換サイトを再現するために,MFIのモデルとしてYumuraらにより作成されたSi92O151H66クラスターを用いた(スキーム2)19)。このモデル中の,ストレートチャネル壁に存在する5員環および6員環サイトを切り出し,モデルとした。それらのモデルにおいて,二つのAl3+の置換位置がZn2+の水素・メタン活性化能におよぼす影響についての知見を得ることを目的として,種々のAl3+の配列パターンのモデルについても検討した。まず,置換した二個のAl3+近傍にZn2+を置いて構造を最適化することで,Zn2+が各サイトにイオン交換されたZn2+の状態を再現した。その後,各構造モデルのZn2+近傍に水素を置き,構造を最適化することで,各サイトにイオン交換されたZn2+の水素活性化能を検討した。その際,種々のサイトを検討したが,以下のモデルを用いた場合に,実験結果を最も良く再現できた。
① MFI中のM7サイトを用いる15,20)。
② そのサイト中にSi原子二個をAl原子に置き換えた(スキーム3および図2)。
③ 二個のAl原子の配置を図に示すように配置した(M7S2モデルと呼称:スキーム3および図2参照)。
④ このサイト上に二価の亜鉛イオンを置き,構造の最適化を行った(図2)。
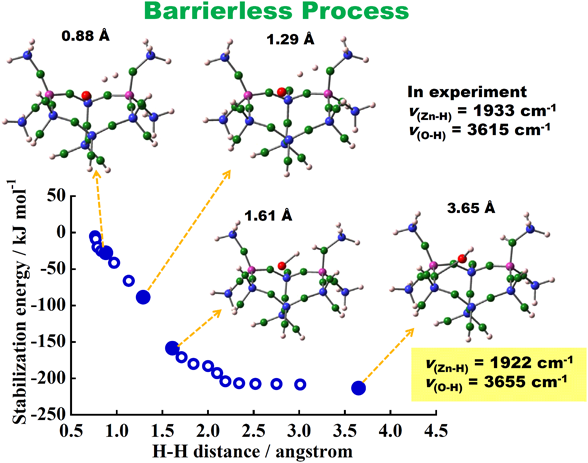
このAl配置を有するモデル,M7S2, に対して,DFT計算法によって,活性化エネルギーを必要としない過程で水素の解離現象を説明できた(図3)。たとえば,赤外線吸収スペクトルの–ZnH, –OHの振動数は実験値と計算値とが良く一致する(図3中のデータ参照)。議論の詳細は文献と解説記事を参照して頂きたい15–18)。
3. ZnMFI, MgMFI上でのメタンの活性化17,21)
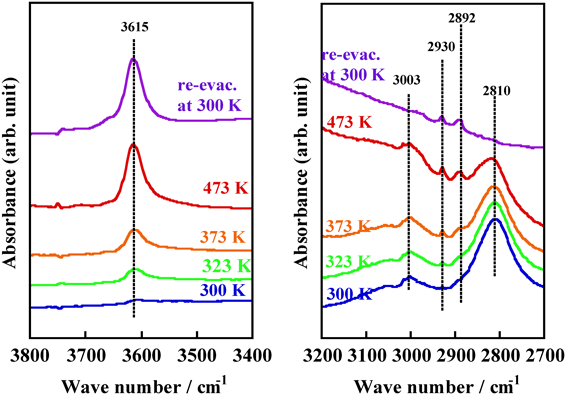
ZnMFI–CH4系においては,ZnMFI–H2系の場合とは少し挙動が異なり,室温ではメタンの解離反応はほとんど起こらない。処理温度が少し高く(323 K以上)なるとメタンが,水素の場合と同様に,不均一結合開裂され,解離吸着種であるZ–OL–[ZnCH3]+種とZ–OL–H+種由来のν(C–H)およびν(O–H)振動が2930, 2892 cm−1と3615 cm−1に観測される(図4:特に,再排気後のスペクトル)17,21)。すなわち,–ZnCH3+種とBrønsted酸点(–OH種)が形成されることがわかった。反応は水素の場合に極めて類似している。
(ここで,ZA and ZBはAl原子を含むゼオライト骨格を意味する。)
次に,水素分子のH–H結合とメタン分子のC–H結合の平均結合エンタルピーは類似した値(それぞれ,436, 412 kJ mol−1)であることを考慮し,ZnMFI中の水素およびメタン解離吸着サイトの関連性を実験的に検討した21)。まず,873 K処理後のZnMFIをメタンガス存在下473 Kで処理し,ZnMFI中の活性点上にメタン解離吸着種を固定化した。続いて,メタンガスを水素ガスに置換し,各温度で熱処理した。水素処理過程についてのIRスペクトル測定の結果を図5に示す。メタン処理していない試料(水素処理により,300 Kでさえ,–ZnH, –OH種が観測される)の場合とは異なり,メタン処理した試料を300 Kで水素ガスにさらした場合には,水素不均一結合開裂種由来のIRバンドは全く観測されない。すなわち,メタン処理後の試料では水素解離反応が生じないことは明らかである。「ZnMFI中の水素解離吸着サイトがメタン解離吸着種によってブロックされた」ことによると解釈するとこの現象をうまく説明できる。水素処理を行う温度が高くなると,水素解離吸着種由来のIRバンドが1933 cm−1に出現し,そのバンド強度は処理温度が高くなるにつれて増加した。一方,メタン解離吸着種由来のIRバンドの強度は処理温度の上昇に伴って減少した。これらの結果は,ZnMFI上で形成したメタン解離吸着種が水素処理過程で水素解離吸着種に置換されたものと解釈できる。以上の結果から,ZnMFI中の同一のサイト上で水素およびメタンの解離反応が起ることを結論した。
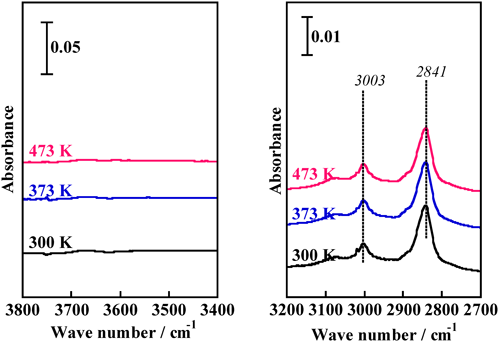
一方,メタン吸着現象についても,MgMFI系と吸着・反応特性の比較を行うために,MgMFI–82試料を用い,メタン活性化が引き起こされるかどうかについて,同様の実験を行った(図6)。300 KでMgMFIをメタンに曝すと,3003, 2841 cm−1に吸収バンドが出現する。これらの吸収バンドは吸着メタンのν3およびν1伸縮振動モードに帰属できる。なお,気体のメタンのν3およびν1伸縮振動モードは,それぞれ,3019, 2917 cm−1に観測される。ただし,Td対称を有するメタンのν1伸縮振動モードは全対称伸縮振動であり,IR不活性である。このモードが,MgMFI–CH4系のIRスペクトルにおいて,2841 cm−1に出現している事実は,Mg2+の強い電場によるメタンとの相互作用の存在を示している22,23)。ゼオライト細孔内でMg2+–CH4間に比較的強い相互作用が存在し,そのことに起因してメタン分子の対称がA1対称から低下したことによると考えることができる。しかし,MgMFI系では上述の二種類のバンド以外のバンド,たとえば,Brønsted酸点種に帰属できるバンド等は全く観測できない。また,ZnMFI系において出現した,(Zn-CH3)種に帰属可能な2930や2892 cm−1のバンドも出現しない。すなわち,Mg–CH3やBrønsted酸点様のメタン解離吸着種によるバンドは生じていないことは明らかである。結果としてMgMFI–CH4系では静電場の効果が相互作用の主なdriving forceであり,ZnMFI–CH4系では静電場の効果に加えて,化学結合が生じていることが結論できる。このように,両系で相互作用に大きな違いがあることが明らかになった。
続いて,ZnMFI中の水素解離吸着サイトはメタン解離吸着サイトとしても機能するという実験事実(図5参照)を基に,M7S2モデルを用いてメタンの解離吸着現象を,計算法を適用して,再現することを試みた17)。その結果,水素分子と同様,メタン分子もM7S2サイトのZn2+上で解離吸着され,Z–OL–[ZnCH3]+種およびZ–OL–H+が形成されることが明らかになった。メタン解離過程での系のエネルギーおよび構造の変化を図7に示す。反応を経て形成したZ–OL–[ZnCH3]+種のνC–H伸縮振動数は3001, 2933 cm−1,Z–OL–H+のνO–H伸縮振動数は3655 cm−1と求まり,実験値(νC–H=2930, 2892 cm−1, νO–H=3615 cm−1)と良い一致を示した(図7)。また,水素解離機構とは異なり,メタン解離機構には遷移状態が存在することがわかった(図7)。このエネルギー走査の結果は,メタン解離過程で外的エネルギーを要するという実験結果に矛盾しない。一連の計算結果から,M7S2モデルのように,Al3+が配置したサイト上にイオン交換されたZn2+上で水素およびメタン分子が解離吸着されることが示唆され,Zn2+の水素・メタン活性能についてAl3+とZn2+との位置関係が重要であることが明らかになった。また,これの結果から,初めて,MFI型ゼオライト中のAl配置に関する情報が得られたことになる。
以上,一連の水素・メタン分子の不均一結合開裂現象は,MFI中にイオン交換されたZn2+上で起ると考えることによってうまく説明できる。ZnMFIは323 K以上でメタンの活性化を促進し,この反応も水素活性化と同一サイト(M7S2サイト)を用いて,現象の説明が可能であることがわかった。
Zn2+イオンによるメタンの活性化に対する特性をMg2+イオンによる活性化との違いを考慮しながら,Zn2+系の特異性(活性化の本質)を解明しようとした。その際,以下の二点に注目した。
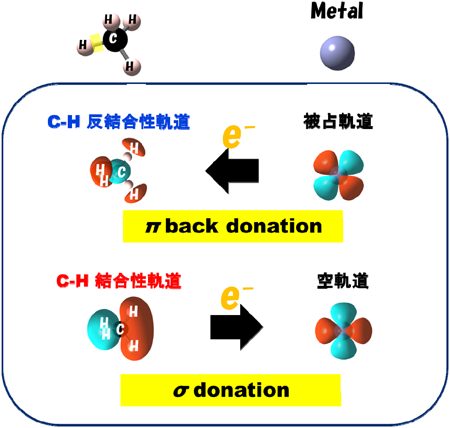
① Zn2+イオンが電子のドナーとして機能するのか,電子のアクセプターとして機能するのかという点に注目した(図8に両モデルを示す)。メタンの分子軌道のHOMOの準位には電子は完全に占められており,メタンの活性化のためには,図9に示すように,LUMOの軌道に金属の軌道から電子を移動するか,HOMOの軌道から金属の軌道に電子を供与するかのどちらかの機構を経て,活性化が進行すると考えられる。
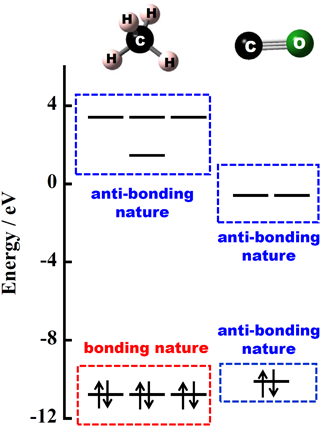
② メタンとCOとの分子軌道図(図9)に着目すると,HOMOとLUMOのエネルギー準位の関係が良く類似していることがわかる。
そこで,COをプローブ分子として利用し,金属イオンの電子状態および金属イオンとCO分子との相互作用の特徴を,吸着CO種のIR測定(室温)により,実験的に検討した。
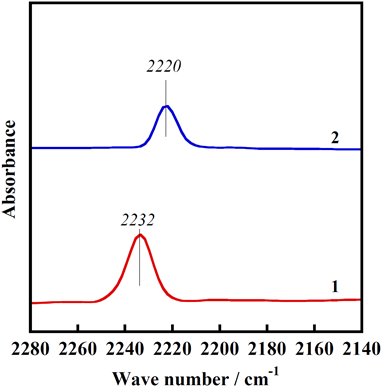
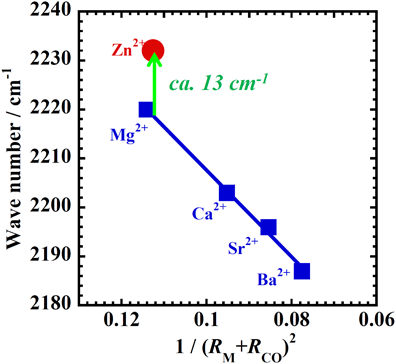
まず,ZnMFI-95およびMgMFI-82試料を873 Kで真空排気し,その後,室温でのCO吸着(低圧領域:強い吸着のみを観測)を行い,その過程のIR測定を行った。その結果を図10に示す。MgMFI系ではMg2+種と相互作用した吸着CO種の伸縮振動バンドが2220 cm−1に観測される。フリーの気体のCO種のバンド(2143 cm−1)から77 cm−1高波数側にシフトしている。この事実は,静電的な相互作用に起因していると解釈できる。実際,Ca2+, Sr2+, Ba2+でイオン交換したMFI型ゼオライトで同様の測定を行うと,図11に示すように電場の強度に対して直線関係が得られ,その解釈が妥当であることを示している。また,前述したMgMFIへのメタン吸着におけるIRデータの解釈(メタンとの相互作用は静電的相互作用によるという結論)と一致する。一方,Mg2+と同じ二価の亜鉛イオンで交換したZnMFI系では吸着CO種のバンドは2232 cm−1に観測される。Mg系と比較して13 cm−1の高波数側へのシフトは,静電的相互作用に加えて,COの反結合性の軌道(5σ)からZn 4s軌道への電子供与の寄与を示している(Mg2+=71 (4) pm, Zn2+=75 (4) pm:括弧内の値は配位数を表す)24)。これらの事実から,MFI中のZn2+は,MFI中のMg2+とは異なり,電子受容体として働き,その結果,メタンのC–H間の不均一結合開裂が引き起こされていると結論できる。この考えを計算によって調べた。その際,M7S2モデルを用いた。その結果,図12に示すように,現象をうまく説明できる。結合エネルギー,赤外線吸収バンドの位置,電子の移動の量およびM–C–Oの結合距離などすべてのデータは,この考え方で合理的に説明できることがわかった。以上のように,MFI中のM7S2サイト上のZn2+は電子受容性の高い電子状態をとり,それがCOの吸着あるいはメタンの活性化に重要な役割をしていると考えることができる。
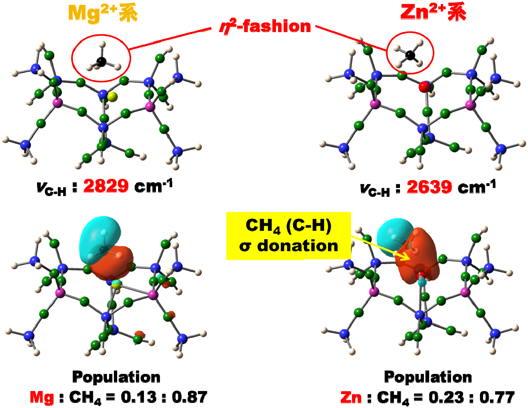
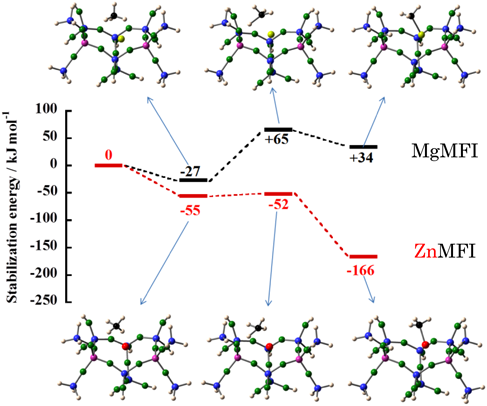
以上の基礎に立脚して,このサイト上でメタンの活性化の機構をZnMFI系とMgMFI系で比較しながら,反応性の違いを示す要因を調べた。上述したように前者では後者より,より高い電子受容性という特性が期待される。まず,それぞれの系でメタンが吸着した状態を図12に示す。二つの系で吸着状態に大きな違いが生ずる。Mg系ではメタン分子のTd対称からのずれは小さいがZn系では大きなずれが見られる。MgMFI系では,吸着メタン種のHOMO軌道はフリーのメタンのHOMOの軌道(1t2)の分子軌道図と殆ど同じであり,また,Mgイオンとの軌道の重なりはほとんど見られない。一方,Zn系ではメタン分子は大きく歪み,同時にZnイオンの軌道との重なりがはっきりと見られる。C–H伸縮振動の波数も,大きく低波数へシフトしている。以上のデータから,CH4–σ (C–H)から亜鉛イオンへの電子の移動が大きいことが明らかである。このようなZnイオンとMgイオンの電子受容能の大きな相違は,LUMO軌道であるZn 4s軌道とMg 3s軌道のエネルギーレベルの差異に起因する。計算ではZn 4s, Mg 3sの準位はそれぞれ,−5.82, −4.29 eVと見積もられる。これらのデータから,上述のメタンおよびCOのエネルギーを考慮すると,Zn2+がMg2+より電子受容性が高いということは明らかである。二つの系でCH4の活性化(不均一結合開裂)過程におけるポテンシャルエネルギー変化を図13にまとめた[初期過程(吸着過程)における,電荷移動について解析を行った結果を図12中に与えてある]。この結果は,Zn2+の場合,−166 kJ/mol(発熱過程),Mg2+の場合,+34 kJ/mol(吸熱過程)となった。また,反応過程における活性化のエネルギーはMg2+の場合,+92 kJ/molと高い障壁であるが,Zn2+の場合には3 kJ/molという小さい障壁となっている。これらの結果は実験事実をうまく説明できることが明らかであり,本系において特異なAl配置をもったサイト上にイオン交換されたZn2+について高い電子受容性が賦与され,その結果,水素やメタンの活性化が促進されることが明らかになった。
以上述べたように,ゼオライト中のM7サイトにおける特異なAl配置(M7S2モデルで表される)は交換イオンに特異な電子状態を賦与し,通常でない電子状態を有する交換金属イオンを生み出していると考えている―新電子状態の創生である。ここで述べた内容は,ゼオライト中のAl配列25–27)と反応性との関連の重要性を議論した初めての研究内容であると認識している。我々は,このような通常の性質とは異なる特性を賦与されたイオンについて“Ultra-ion”と提案している。「はじめに」のところで述べた,ゼオライト中のCu+もそのようなイオンの例であると理解している。今後,ここで提案しているモデルについてさらに実験的な確証を得ること,また,提案しているような視点から細孔物質をデザインできればと思うとともに,そういう特性の創生をめざした研究の発展を期待している。ゼオライトには,まだまだ新しい興味深い現象が潜んでいる。
謝辞Acknowledgments
本研究は京都工芸繊維大学の小林久芳教授,湯村尚史准教授との共同研究である。特に,本系への計算法の適用を行う上で,御指導いただくとともに,議論を行っていただいた。また,研究の一部は科学研究費の援助および学術振興会のDC1(織田晃および鳥越裕恵)の援助を得ることによって行うことができた。さらに,これまで多くの研究者や院生諸君の協力を得て本研究を行うことができた。特に,研究室の大久保貴広准教授と板谷篤司特任助教(現新潟大学工学部)には,実験から研究の議論をとおして多大のご貢献を頂いた。以上,ここに紙面をお借りして感謝の意を表したい。
引用文献References
1) K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka, and H. Kitagawa, J. Am. Chem. Soc., 136, 1864–1871 (2014).
2) S. Shan, V. Petkov, L. Yang, J. Luo, P. Joseph, D. Mayzel, B. Prasai, L. Wang, M. Engelhard, and C.-J. Xhong, J. Am. Chem. Soc., 136, 7140–7151 (2014).
3) E. C. Tyo, A. W. Castleman, Jr., A. C. Reber, and S. N. Khanna, J. Phys. Chem. C, 115, 16797–16802 (2011).
4) A. W. Castleman, Jr., J. Phys. Chem. Lett., 2, 1062–1069 (2011).
5) M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S.-i. Mikuriya, and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 1272–1273.
6) Y. Kuroda, Y. Yoshikawa, R. Kumashiro, and M. Nagao, J. Phys. Chem. B, 101, 6497–6503 (1997).
7) A. Itadanim M. Tanamka, T. Mori, M. Nagao, H. Kobayashi, and Y. Kuroda, J. Phys. Chem. C, 111, 12011–12023 (2007).
8) Y. Kuroda, S.-i. Konno, K. Morimoto, and Y. Yoshikawa, J. Chem. Soc., Chem. Commun., 1993, 18–20.
9) R. Kumashiro, Y. Kuroda, and M. Nagao, J. Phys. Chem., 103, 89–96 (1999).
10) Y. Kuroda, Y. Yoshikawa, S. Emura, R. Kumashiro, and M. Nagao, J. Phys. Chem., 103, 2155–2164 (1997).
11) A. Itadani, M. Tanaka, T. Mori, H. Torigoe, H. Kobayashi, and Y. Kuroda, J. Phys. Chem. Lett., 1, 2385–2390 (2010).
12) T. Yumura, H. Yamashita, H. Torigoe, H. Kobayashi, and Y. Kuroda, Phys. Chem. Chem. Phys., 12, 2392–2400 (2010).
13) H. Torigoe, T. Mori, K. Fujie, T. Ohkubo, A. Itadani, K. Gotoh, H. Ishida, H. Yamashita, T. Yumura, H. Kobayashi, and Y. Kuroda, J. Phys. Chem. Lett., 1, 2642–2650 (2010).
14) Y. Ono, Cat. Rev. Sci. Eng., 34, 179–226 (1992).
15) A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi, and Y. Kuorda, Angew. Chem. Int. Ed., 57, 7719–7723 (2012).
16) A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi, and Y. Kuorda, J. Am. Chem. Soc., 135, 18481–18489 (2013).
17) A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi, and Y. Kuroda, J. Phys. Chem. C., 118, 15234–15241 (2014).
18) 織田晃,鳥越裕恵,黒田泰重,触媒,55, 352–357 (2013).
19) T. Yumura, M. Takeuchi, H. Torigoe, H. Kobayashi, and Y. Kuroda, Inorg. Chem., 48, 508–517 (2009).
20) D. Nachtigallova, P. Nachtigall, M. Sierka, and J. Sauer, Phys. Chem. Chem. Phys., 1, 2019–2026 (1999).
21) A. Oda, H. Torigoe, A. Itadani, T. Ohkubo, T. Yumura, H. Kobayashi, and Y. Kuorda, J. Phys. Chem. C., 117, 19525–19534 (2013).
22) T. Yamazaki, I. Watanuki, S. Ozawa, and Y. Ogino, Langmuir, 4, 433–438 (1988).
23) T. Yamazaki, K. Hasegawa, K. Honma, and S. Ozawa, Phys. Chem. Chem. Phys., 3, 2686–2691 (2001).
24) R. D. Shannon, Acta Cryst., A32, 751–761 (1976).
25) J. A. van Bokhoven, T.-L. Lee, M. Drakopoulos, C. Lamberti, S. Thiess, and J. Zegenhagen, Nature Mater., 7, 551–555 (2008).
26) A. Janda, and A. T. Bell, J. Am. Chem. Soc., 135, 19193–19207 (2013).
27) A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu, and J. A. Lercher, J. Am. Chem. Soc., 136, 8296–8306 (2014).