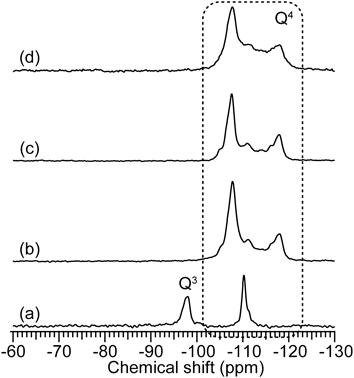
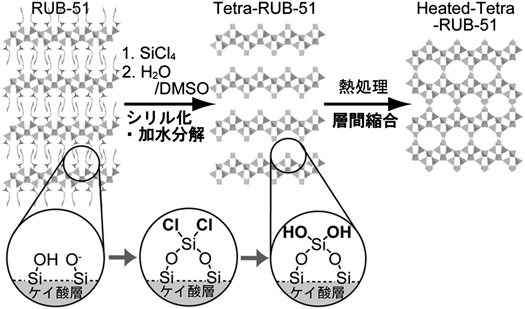
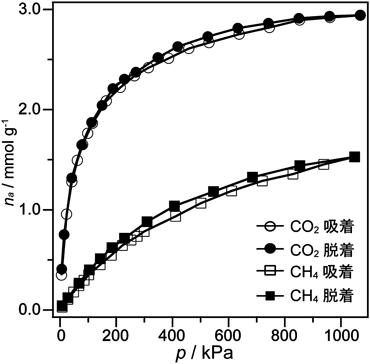
21) a) Y. Oumi, T. Takeoka, T. Ikeda, T. Yokoyama, T. Sano, New J. Chem., 31, 593 (2007); b) T. Ikeda, Y. Oumi, T. Takeoka, T. Yokoyama, T. Sano, T. Hanaoka, Microporous Mesoporous Mater., 110, 488 (2008); c) Y. Oumi, K. Takagi, T. Ikeda, H. Sasaki, T. Yokoyama, T. Sano, J. Porous Mater., 16, 641 (2009); d) T. Moteki, W. Chaikittisilp, Y. Sakamoto, A. Shimojima, T. Okubo, Chem. Mater., 23, 3564 (2011).
22) G. Lagaly, K. Beneke, A. Weiss, Am. Mineral., 60, 650 (1975).
27) a) S. Inagaki, T. Yokoi, Y. Kubota, T. Tatsumi, Chem. Commun., 5188 (2007); b) P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki, T. Tatsumi, J. Am. Chem. Soc., 130, 8178 (2008); c) S. Inagaki, T. Tatsumi, Chem. Commun., 2583 (2009); d) J. F. Ruan, P. Wu, B. Slater, Z. L. Zhao, L. L. Wu, O. Terasaki, Chem. Mater., 21, 2904 (2009); e) H. Gies, U. Muller, B. Yilmaz, T. Tatsumi, B. Xie, F. S. Xiao, X. H. Bao, W. P. Zhang, D. De Vos, Chem. Mater., 23, 2545 (2011); f) S. Inagaki, H. Imai, S. Tsujiuchi, H. Yakushiji, T. Yokoi, T. Tatsumi, Microporous Mesoporous Mater., 142, 354 (2011); g) B. Tijsebaert, M. Henry, H. Gies, F.-S. Xiao, W. Zhang, X. Bao, H. Imai, T. Tatsumi, U. Müller, B. Yilmaz, P. Jacobs, D. D. Vos, J. Catal., 282, 47 (2011); h) F.-S. Xiao, B. Xie, H. Zhang, L. Wang, X. Meng, W. Zhang, X. Bao, B. Yilmaz, U. Müller, H. Gies, H. Imai, T. Tatsumi, D. De Vos, ChemCatChem, 3, 1442 (2011); i) H. Gies, U. Muller, B. Yilmaz, M. Feyen, T. Tatsumi, H. Imai, H. Y. Zhang, B. Xie, F. S. Xiao, X. H. Bao, W. P. Zhang, T. De Baerdemaeker, D. De Vos, Chem. Mater., 24, 1536 (2012); j) B. Yilmaz, U. Muller, M. Feyen, H. Zhang, F.-S. Xiao, T. De Baerdemaeker, B. Tijsebaert, P. Jacobs, D. De Vos, W. Zhang, X. Bao, H. Imai, T. Tatsumi, H. Gies, Chem. Commun., 48, 11549 (2012); k) J. G. Jiang, L. L. Jia, B. T. Yang, H. Xu, P. Wu, Chem. Mater., 25, 4710 (2013); l) H. Xu, B. Yang, J.-g. Jiang, L. Jia, M. He, P. Wu, Microporous Mesoporous Mater., 169, 88 (2013).