MOFの細孔空間を活用した高分子合成Polymer Synthesis in Nanochannels of MOF
京都大学大学院工学研究科・JST-CRESTGraduate School of Engineering, Kyoto University, JST-CREST ◇ 〒606-8510 京都市西京区京都大学桂
受理日:2014年6月9日Accepted: June 9, 2014
発行日:2014年9月10日Published: September 10, 2014
金属イオンと有機配位子との自己集合によって得られる多孔性金属錯体(MOF)はテーラーメイド設計が可能な規則性ナノ空間を骨格内部に有している。このような有機―無機複合材料が形成するナノ細孔のサイズ,形状,表面機能を合理的に設計することで,革新的な高分子制御場としての利用が可能になる。すなわち,MOFのナノ空間内で重合を行うことで,高分子の一次構造(分子量,立体規則性,反応位置など)や高次集積構造(形状,周期性,配向など)を精密に制御するだけではなく,今まで合成が望まれていたにも関わらず,通常法では全く不可能だった高分子材料の合成や,潜在的に有していた機能性を発現する超構造高分子集積体の構築ができるようになってきた。
Metal-Organic Frameworks (MOFs) composed of transition metal ions and bridging organic ligands have been extensively studied. The characteristics of MOFs are highly regular channel structures, controllable channel sizes approximating molecular dimensions, designable surface potentials and functionality, and flexible frameworks responsive to guest molecules. Owing to these advantages, successful applications of MOFs range from molecular storage/separation to heterogeneous catalysts. In particular, use of their regulated and tunable nanochannels for a field of polymerization allows multi-level controls of polymerization. This account focuses on recent progress in polymerization using the nanochannels of MOFs, and demonstrates that this methodology for polymer synthesis is a promising strategy for the controlling polymer primary structures, creation of precision polymer assembly, and the design of well-defined nanohybrid materials.
キーワード:高分子合成;多孔性金属錯体;ナノチャネル;一次構造;高分子集積
Key words: Polymerization; Metal organic framework; Nanochannel; Primary structure; Polymer arrangement
© 2014 ゼオライト学会© 2014 Japan Association of Zeolite
高分子は金属,セラミックスと並ぶ三大材料であり,我々の生活を支えるキーマテリアルとなっている。合成高分子の利用分野は,この材料が持つ軽量性や高い加工性などの特徴から年々拡大し続けており,高い機能性を示す高分子材料の開発が強く望まれている。ただ,我々が高分子材料を合成する際,一般的には,フラスコや反応釜といったマクロスケールの反応容器を用いる。この場合,大量に安価な高分子材料を提供できるという点では良いが,得られる高分子鎖は必然的に絡み合ってしまい,その構造制御も困難なことが多い。もし,目的とする高分子鎖に見合ったスケールの空間を反応容器として用いることができれば,モノマーの配向,位置,距離,電子状態などを巧に制御できるはずである。すなわち,容器の空間自体が高分子合成反応(重合反応)に大きな影響を及ぼすことが期待され,得られる高分子の一次構造や集積状態を精密に制御することが可能になる。このような背景のもと,骨格内に多数のナノ細孔を有する物質群(ゼオライト,粘土鉱物,有機ホストなど)を重合反応場として利用する試みが行われ,いくつかの高分子材料の合成制御が行われてきた1–4)。しかし,近年の高分子材料の発展と多様性を考えると,様々な種類の高分子を目的に応じた構造,集積様式で合理的に得るシステムの開発が急務となっている。このような観点から,重合反応場として用いる空間の構造(サイズ,形状,表面機能性など)を自在に設計することができれば,その空間情報に応じた高分子材料を効率よく得るシステムが構築できるはずである。本解説では,近年,新しいナノ空間材料として注目を集めている多孔性金属錯体(MOF)を用いることで,通常法では困難,もしくは全く不可能な高分子材料の合成や,超精密な高分子集積体の構築が可能になることを最近の研究例を挙げて紹介する2,5,6)。
金属イオンと有機配位子との自己集合反応により,ナノサイズ(0.4~3 nm)の規則的な空間を有するMOFが合成できる(図1)7–9)。この材料では,金属イオンと有機配位子の組み合わせが原理的に無限に存在することから,細孔構造の設計に関して無限の可能性を有する。使用するビルディングブロックを決定すれば,その誘導体配位子を用いることで,空間のサイズ制御や官能基の導入が可能になる。またビルディングブロックの電子構造をチューニングすることで,単なる空間構造のみならず電子物性,化学反応性の付与も可能になる。その上,骨格構造が超分子構造によって構築されるため,種々の外部刺激(光,熱,電場,磁場,ゲスト分子など)によって,空間構造を動的に変化させることもできる。つまり,金属イオンと配位子の組み合わせを適切に考慮することで,自分の欲しい空間構造や機能をテーラーメイド創製できるという有用性がある。このような特徴から,近年ではガス吸蔵や分離,輸送材料などの応用が検討されており,錯体化学のみならず幅広い分野の研究者から注目を集めている10–13)。高分子の観点からみると,MOFの細孔は一般的な高分子の鎖がちょうど一本程度で入る大きさであり,このような機能性細孔を重合反応場として用いれば,空間構造が重合反応に大きな影響を与えることは間違いない(図2)2,5,6)。MOFのナノ空間の特徴を利用することで,重合反応場としてどのような効果が期待されるのかを以下に示す。
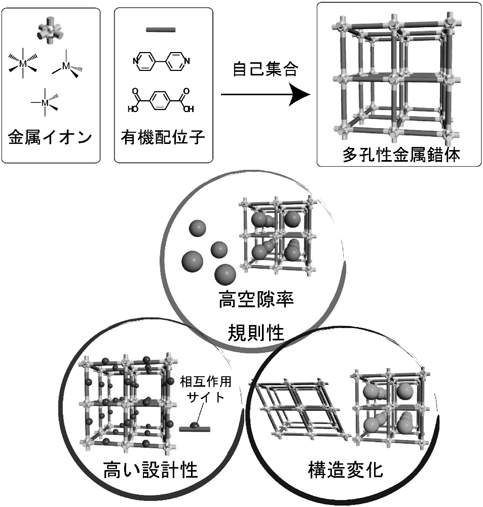
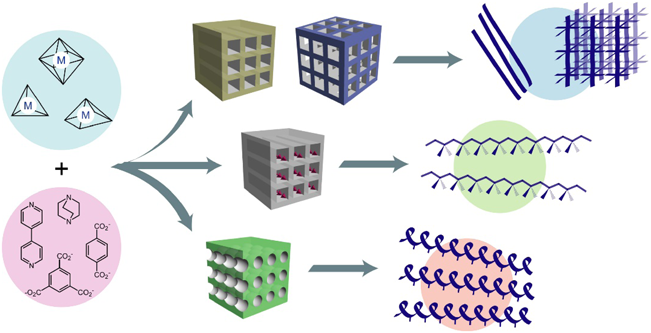
金属イオンと有機配位子との配位結合により合成される過程において,溶液内での自己集合・自己組織化が何度も繰り返されることで,高い結晶性を有するMOFが生み出される。これにより,MOF粒子中では細孔サイズや形状が全く均一な規則性空間が構築される。このような空間内にモノマー分子を導入すると,通常のバルクや溶液中とは異なり,モノマーの位置や向きが規則的に配列した特異な状態を作り出すことができる。モノマーの配列が規制されることで,生成高分子の構造にも規則性を付与することが期待でき,ランダムなアモルファス状態を示す一般的な高分子材料とは一線を画する材料となる。また,空間の次元性も規制できることから,細孔内で高分子鎖の配向構造は完璧に制御できる。一般的にMOFはキレート溶液などで処理することで,容易に構造を壊すことができ,高分子鎖の配向状態が保たれた機能性材料も構築できる。
MOFの配位子を設計することで細孔のサイズや形状の制御が可能になる。これにより,導入モノマーの配列状態のチューニングや高分子成長過程における反応の方向性などを制御できる。細孔サイズは導入モノマーの運動性に直接関わってくるところであり,サイズが小さくなれば,細孔壁との相互作用が強くなり,モノマーの運動性(≒反応性)は下がる。しかし,モノマーの配向や反応の選択性は厳密に制御できる方向に進むので,これらのバランスを上手く取ることが望まれる。細孔形状に関しても,モノマーの位置や反応方向を規制する重要なファクターとなる。例えば,不斉配位子を用いることで,キラルな環境を作り出せ14),細孔内で重合を行うことで,不斉選択重合やらせん高分子の合理的な合成も可能になる。
そもそもMOFの骨格は遷移金属イオンで構成されているものが多く,フレームワーク内の金属イオンサイトを活用することで,様々な重合反応を進行させることができる。この際,配位不飽和な金属イオンサイトを構築することが重要で,モノマーと相互作用させることで,配位重合,酸化重合などを効果的に触媒することができる。また,配位子の方にも水素結合サイトや反応サイトを付与することができるので,これにより,モノマーの配向や序列の制御,および高分子架橋の制御などが可能になる。ナノチャネル内に周期的にこのようなサイトを導入することで,得られる高分子の立体規則性や定序性,架橋規則性の制御を達成できる。
MOFは非共有型の配位結合により骨格が構築されており,ゼオライトや活性炭では示さないような動的細孔挙動を示す場合がある9,15)。この特性を利用すれば,モノマーの特異的な認識や配向が可能になり,精密構造制御が可能な重合系に発展する可能性がある。また,重合途中に様々な物理的外部刺激を与えることで,空間の構造を変化させ,生成高分子の分子量制御やブロック共重合体の合成などが達成できる。重合後にMOF構造を変化させることで,高分子の取り出しが容易に行えることも期待でき,何度も利用可能な重合制御場としての利用も視野に入る。
ラジカル重合は最も古典的な重合法の一つで,古くから研究,開発が行われており,現在の産業においても主要な役割を演じている16,17)。しかし,反応性の高いラジカル種を介した連鎖機構で反応が進むため,その制御が極めて難しく,得られる高分子の分子量,立体規則性,共重合定序性,反応位置などの一次構造制御はいまだに困難である。筆者らは主に1次元チャネルを有する[M2(L)2(ted)]n(M=Cu2+ or Zn2+, L=テレフタル酸系ジカルボキシレート,ted=triethylenediamine)の細孔中で種々のビニルモノマーのラジカル重合を行った。
本MOF内にスチレンやメタクリル酸メチルなどのビニルモノマーを導入後,ラジカル開始剤とともに加熱することで重合を行った。重合途中の,成長ラジカルの様子をESR測定により観測した18,19)。その結果,通常のバルクや溶液中での重合に比べて,成長ラジカルがはるかに高濃度,長寿命であることが明らかになった。得られた高分子の分子量を測定すると,数万程度の高分子量体であることが分かり,分子量分布が通常法で得られたものに比べて狭くなった。これは,ナノ細孔中の成長ラジカルが単分子鎖の状態で保護され,成長反応以外の副反応が抑制されたため,ナノ細孔中での重合反応がリビング重合的に進行したことを示している。すなわち,MOFのナノ空間内では反応性が高い成長ラジカルを制御することが可能になることが分かった。
MOFのナノ空間内で得られたビニル高分子の立体規則性の変化について検討した19–21)。その結果,通常のバルクや溶液重合とは立体規則性が異なる高分子が得られることがわかった。配位子を変えることでMOFの細孔サイズを小さくすると,側鎖が同じ方向に突き出したメソユニットの割合が増えていくということが確認された19)。これは狭い細孔内では,立体的に嵩高いラセモユニットを取ることが難しく,構造的に小さいメソ構造を取りやすくなるためと説明できる。また,不飽和金属イオンサイトを用いて空間内でモノマーの配向を規制することや20),配位子に種々の置換基を導入することでMOF細孔の形状を制御することでも21),得られる高分子の立体規則性に大きな影響を及ぼすことがわった(図3)。これにより,ラジカル重合では達成が困難なメソユニットが豊富な高分子(ポリメタクリル酸メチルやポリ酢酸ビニル)の合成にも成功した。
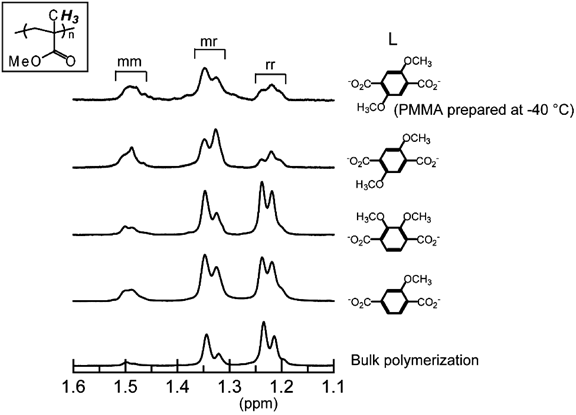
mm=isotactic triad, mr=heterotactic triad, and rr=syndiotactic triad.
数種類のモノマー混合物のラジカル重合においては,ほとんどの場合,モノマー同士がランダムに重合し,定序性を持たない共重合体を生成する。これに対し,MOFの細孔で共重合を行うと,通常のバルクや溶液重合と比べ,モノマーの反応性が変化することが明らかとなった22)。これは,ナノサイズの細孔内でモノマーの運動性が制限されるためと考えられ,共重合におけるモノマー組成を制御できる可能性を示している。細孔の周期性を意識したモノマー配列を行えば,これまで不可能であった定序性の精密制御が可能になるかもしれない。
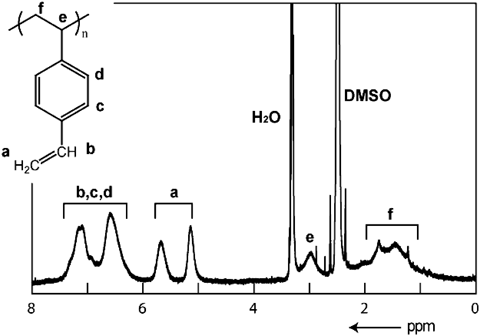
複数の反応サイトを有するジビニルベンゼン(DVB)はラジカル重合において架橋剤として用いられ,不溶性架橋高分子やゲル材料を合成する際に重要な役割を演じる。このモノマーを[M2(tp)2(ted)]n(tp=terephthalate)の一次元ナノ細孔中で重合を行った23)。すると,M=Zn2+のナノチャネル中ではDVBの重合が高効率で進行し,可溶性の高分子が得られることがわかった。種々のスペクトル測定からDVBの片方のビニル基だけが選択的に重合し,直鎖状の高分子が得られたということが分かった(図4)。一方,同じ細孔サイズを有するM=Cu2+の細孔中では全く重合反応は進行しなかった。この重合反応性の違いは錯体の骨格柔軟性に起因することがわかり,XRPD測定から亜鉛系錯体の細孔構造は動的に変化し,細孔内で重合可能なDVBの近接配置ができることが分かった。DVBの二つのビニル基の反応性は全く等価であり,片方だけを選択して重合することは不可能であったが,MOFのナノ空間を反応場にすることで初めてサイト選択的に重合を進行させることに成功した。
多孔性金属錯体の細孔を用いることで,ビニル高分子の集積制御も可能になる。実際,種々のMOF中で高分子を合成し,キレート剤で処理することで高分子の単離を行うと,元々のホストMOFのモルフォロジーを保持した高分子集積型微粒子が得られた24–27)。これにより,立方体状,ロッド状,六角柱状の高分子微粒子が効率よく構築できることがわかり,非球状の高分子微粒子を作り出す合理的な手法として期待される。
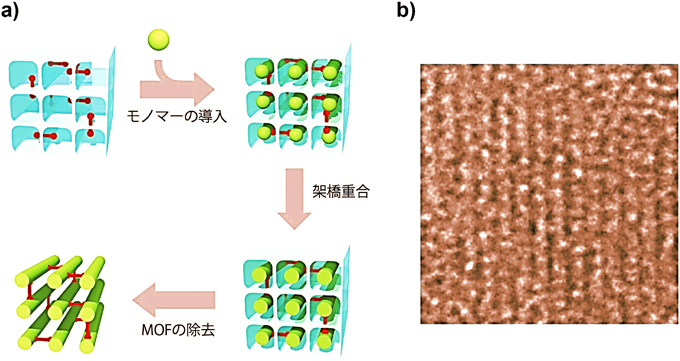
最近,MOFの一次元ナノ細孔内で高分子の合成と架橋を同時に行い,反応後にホストを除去することで,一本一本の高分子鎖が同じ方向に整列した新しい高分子材料を開発した(図5)28)。ジビニル型テレフタレートを[Cu2(tp)2(ted)]n骨格に一部組み込み,導入されたビニルモノマーとMOFとの間で架橋重合後,ホストMOFの除去を行った。単離した高分子のXRPD測定を行ったところ,高分子鎖のパッキングに由来する回折ピークが確認された。そこで,高分解能TEM測定を行うと,高分子の鎖が一次元的に整列している像が確認された。このような配向状態は高分子鎖同士が架橋されているために安定で,有機溶媒処理や高温処理しても,その配向構造が保たれる。また,ここで得られた高分子の比重測定を行うと,通常のバルク状態に比べて高い値を示し,高分子鎖が密にパッキングしていることが示された。つまり,この手法により,単なる汎用プラスチック材料が,耐溶剤・耐熱性を備えた高強度エンジニアリングプラスチックとして生まれ変わる可能性がある。
魅力的な電子・光機能を示すπ-共役高分子の構造を制御しながら合成ができれば,これらの持つ機能を合理的にチューニングすることができる。一般的に,π-共役高分子は種々の均一系触媒を使った溶液中で合成されることが多い。MOFに関しても,細孔表面に特異な活性点や触媒作用点を規則的に配置することが可能であり29,30),このような触媒的性質を示す細孔を用いて,π-共役高分子の合成制御を行うことができる。
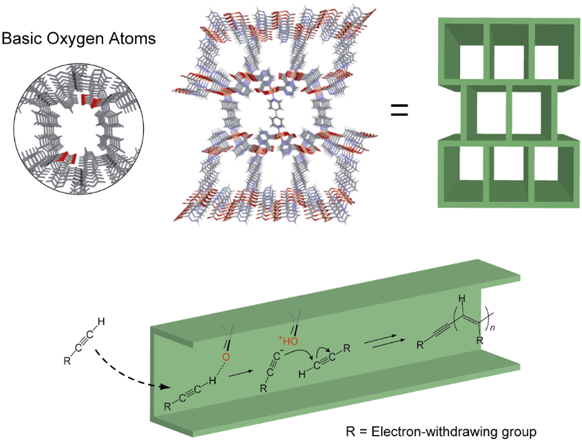
MOFの細孔表面は様々な有機官能基で修飾することが可能であり,このような表面官能基とゲスト分子(ガスや溶媒)との間で相互作用が働くことで,ゲスト選択的な吸着挙動が観測される。例えば,細孔表面にカルボキシレート基が突き出した構造を持つピラードレイヤー型MOFは,アセチレンの末端水素と水素結合することで,細孔内に高密度でアセチレンを充填できることが報告されている31)。このような細孔内に,電子受容性基が結合した一置換型酸性アセチレン(メチルプロピオレートやエチニルピリジンなど)を導入すると,自発的に重合反応が進行することがわかった(図6)32)。この系では,二置換アセチレンや酸性度の低い一置換アセチレンでは重合が進行しなかったことから,吸着モノマーと細孔表面との強い水素結合により生じる活性アセチリドが開始剤となり,重合が進行することが示唆された。興味深いことに,MOFの狭い一次元空間で重合が進行するため,トランス付加が優先されたポリ置換アセチレンが生成することが分かった。
骨格中にFe(III)を有する二次元層状MOFのレイヤー間でピロールの酸化重合を行った33)。キレート水溶液中でホスト錯体を除去して得られたポリピロールにおいて,ピロールπ環の積層構造に基づく構造秩序が存在することが示唆された。つまり,二次元レイヤー状の錯体空間を鋳型とすることで,二次元的に配向したポリピロールが得られることが分かった。興味深いことに,得られたポリピロールシートの導電性を測定すると,シートに対して平行方向の導電性が垂直方向に比べて20倍高いという異方性を示すことが明らかになった。
三次元的に連結された細孔を有するMOFを鋳型とすることでも,ポリピロールの配向制御ができる34)。このようなMOF中で合成・単離後,得られたポリピロールのガス吸着測定を行った。その結果,通常のバルク状態で合成したポリピロールは全くガス吸着挙動を示さなかったのに対し,複合体から単離したポリピロールは確かな吸着挙動を示し,多孔性構造を有することが分かった。
有機高分子のみにとどまらず,金属酸化物に代表される無機高分子に関しても,多孔性錯体のナノ空間を使って構造制御ができる。例えば,シリカを合成する方法の一つにゾル–ゲル法がある。これはアルコキシシランを穏和な条件で加水分解・重縮合するという簡便な手法であり,ガラスや有機–無機ナノハイブリッド材料の作製など幅広く用いられている。MOFが形成する空間内でゾル–ゲル反応を行うことで,サブナノナノレベルで制御されたシリカの合成が可能になった35,36)。1 nm以下の細孔系を有するMOFのチャネル中にテトラメトキシシランを導入し,加水分解および重縮合反応を行うことで,細孔内でシリカの合成を行った。得られた複合体の29Si NMR測定から,ナノ細孔の規制を強く受けながらゲル化し,表面シラノール基が多数残存する極微サイズのシリカ粒子が生成することが分かった35)。興味深いことに,この低次元ナノ構造は空気中では1年以上安定で,水で処理してもほとんど影響を受けないことが分かった。また,シリカは高温処理することで結晶化し,石英やクリストバライトなどに転移することが知られているが,ここで得られたシリカは,サイズが微小化しているため,クリストバライト相への結晶化温度がバルク状態に比べて700°C近くも低下することがわかった。
ここで得られた複合体の吸着測定を行うと,シリカ表面のシラノール基が示す親水性により,ホスト単体に比べて水の吸着が促進される結果が得られた36)。極めて微量のシリカ微粒子でも細孔内に存在すると有効に機能することが分かり,多孔性錯体の構造を変えなくとも,このような無機高分子を“ドープ”するだけで,効果的な機能変換ができることを明らかにした。シリカだけではなく,チタニアに関してもMOF空間内で合成できることが分かり,光応答性の吸着剤として有用な機能を示すことが分かってきている37)。
多孔性金属錯体の設計可能なナノ細孔を重合反応場として用いることで,従来法では困難,あるいは不可能な高分子を合成でき,高分子構造の精密制御が可能になることを示した。このようなMOF空間は高分子の合成制御場としてだけではなく,空間内に高分子鎖を精密に配列することで,拘束高分子による特異な機能を発現するナノ複合材料としても期待できる24,38–41)。単純にナノ空間と言うと“有限”な場のイメージだが,機能性高分子を創成するという観点からは“無限”の可能性を秘めている。現在の産業では巨大スケールの反応容器を用い,大量・安価に高分子材料を提供することに重点を置いているが,新興国の台頭もあり,国内生産量は減少の一途をたどっている。近い将来,ナノスケールの工場から多くの未来材料が生まれる日を夢見ている。
1) C. M. Paleos, Polymerization in Organized Media, Gordon & Breach: New York (1992).
2) T. Uemura and S. Kitagawa in Synthesis of Polymers: New Structures and Methods, Wiley-VCH, Weinheim, p. 1011 (2012).
3) M. Miyata in Comprehensive Supramolecular Chemistry, Vol. 10, Pergamon, Oxford p. 557 (1996).
4) D. J. Cardin, Adv. Mater., 14, 553 (2002).
5) T. Uemura, S. Horike, and S. Kitagawa, Chem. Asian J., 1, 36 (2006).
6) T. Uemura, N. Yanai, and S. Kitagawa, Chem. Soc. Rev., 38, 1228 (2009).
7) H. Furukawa, K. E. Cordova, M. O’Keeffe, and O. M. Yaghi, Science, 341, 1230444 (2013).
8) S. Kitagawa, R. Kitaura, and S.-i. Noro, Angew. Chem. Int. Ed., 43, 2334 (2004).
9) G. Férey and C. Serre, Chem. Soc. Rev., 38, 138 (2009).
10) J.-R. Li, J. Sculley, and H.-C. Zhou, Chem. Rev., 112, 869 (2012).
11) L. J. Murray, M. Dincă, and J. R. Long, Chem. Soc. Rev., 38, 1294 (2009).
12) M. D. Allendorf, C. A. Bauer, R. K. Bhakta, and R. J. T. Houk, R. Chem. Soc. Rev., 38, 1330 (2009).
13) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur, and C. Serre, Angew. Chem. Int. Ed., 49, 6260 (2010).
14) M. Yoon, R. Srirambalaji, and K. Kim, Chem. Rev., 112, 1196 (2012).
15) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 34, 109 (2005).
16) G. Moad and D. H. Solomon, The Chemistry of Radical Polymerization, 2nd ed, Elsevier, Oxford (2006).
17) K. Matyjaszewski and T. P. Davis, Handbook of Radical Polymerization, Wiley-Interscience, Hoboken (2002).
18) T. Uemura, K. Kitagawa, S. Horike, T. Kawamura, S. Kitagawa, M. Mizuno, and K. Endo, Chem. Commun., 5968 (2005).
19) T. Uemura, Y. Ono, K. Kitagawa, and S. Kitagawa, Macromolecules, 41, 87 (2008).
20) T. Uemura, N. Uchida, M. Higuchi, and S. Kitagawa, Macromolecules, 44, 2693 (2011).
21) T. Uemura, Y. Ono, Y. Hijikata, and S. Kitagawa, J. Am. Chem. Soc. 132, 4917 (2010).
22) T. Uemura, Y. Ono, and S. Kitagawa, Chem. Lett. 37, 616 (2008).
23) T. Uemura, D. Hiramatsu, Y. Kubota, M. Takata, and S. Kitagawa, Angew. Chem. Int. Ed., 46, 4987 (2007).
24) T. Uemura, N. Uchida, A. Asano, A. Saeki, S. Seki, M. Tsujimoto, S. Isoda, and S. Kitagawa, J. Am. Chem. Soc., 134, 8360 (2012).
25) T. Uemura, T. Kaseda, and S. Kitagawa, Chem. Mater., 25, 3772 (2013).
26) T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado, and K. Sada, J. Am. Chem. Soc., 135, 5427 (2013).
27) Q.-X. Wang and C.-Y. Zhang, Macromol. Rapid Commun., 32, 1610 (2011).
28) G. Distefano, H. Suzuki, M. Tsujimoto, S. Isoda, S. Bracco, A. Comotti, P. Sozzani, T. Uemura, and S. Kitagawa, Nature Chem., 5, 335 (2013).
29) B. Kesanli and W. Lin, Coord. Chem. Rev., 246, 305 (2003).
30) Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen, and J. T. Hupp, Chem. Soc. Rev. 38, 1450 (2009).
31) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe, and Y. Mita, Nature, 436, 238 (2005).
32) T. Uemura, R. Kitaura, Y. Ohta, M. Nagaoka, and S. Kitagawa, Angew. Chem. Int. Ed., 45, 4112 (2006).
33) N. Yanai, T. Uemura, M. Ohba, Y. Kadowaki, M. Maesato, M. Takenaka, S. Nishitsuji, H. Hasegawa, and S. Kitagawa, Angew. Chem. Int. Ed., 47, 9883 (2008).
34) T. Uemura, Y. Kadowaki, N. Yanai, and S. Kitagawa, Chem. Mater. 21, 4096 (2009).
35) T. Uemura, D. Hiramatsu, K. Yoshida, S. Isoda, and S. Kitagawa, J. Am. Chem. Soc., 130, 9216 (2008).
36) T. Uemura, Y. Kadowaki, C. R. Kim, T. Fukushima, D. Hiramatsu, and S. Kitagawa, Chem. Mater., 23, 1736 (2011).
37) C. R. Kim, T. Uemura, and S. Kitagawa, Micropor. Mesopor. Mater., 195, 31 (2014).
38) T. Uemura, S. Horike, K. Kitagawa, M. Mizuno, K. Endo, S. Bracco, A. Comotti, P. Sozzani, M. Nagaoka, and S. Kitagawa, J. Am. Chem. Soc., 130, 6781 (2008).
39) T. Uemura, N. Yanai, S. Watanabe, H. Tanaka, R. Numaguchi, M. T. Miyahara, Y. Ohta, M. Nagaoka, and S. Kitagawa, Nature Commun., 1, 83 (2010).
40) N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Tanaka, M. Mizuno, T. Uemura, and S. Kitagawa, Nature Mater., 10, 787 (2011).
41) Y. Ikezoe, G. Washino, T. Uemura, S. Kitagawa, and H. Matsui, Nature Mater., 11, 1081 (2012).
This page was created on 2017-03-27T16:35:12.624+09:00
This page was last modified on
このサイトは(株)国際文献社によって運用されています。