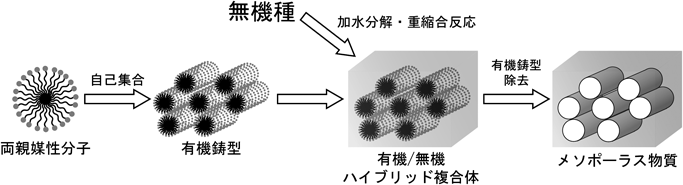
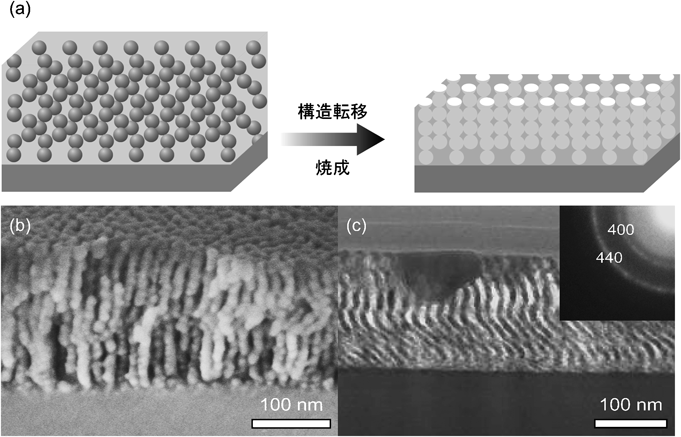
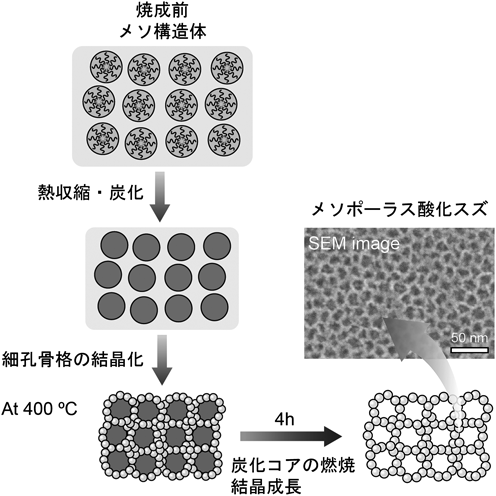
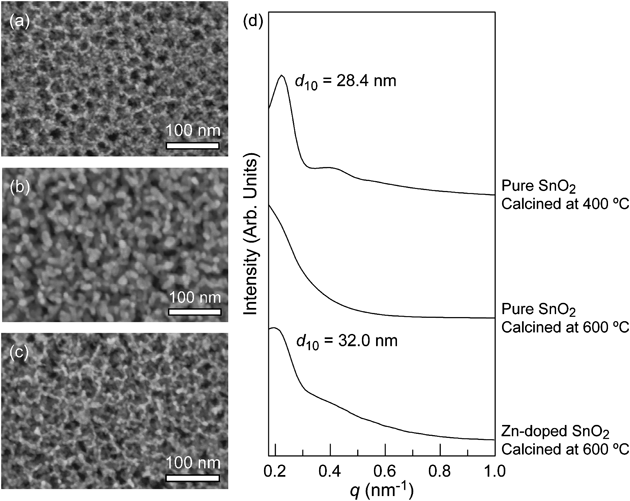
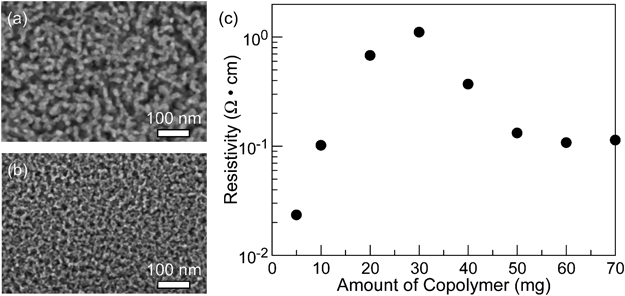
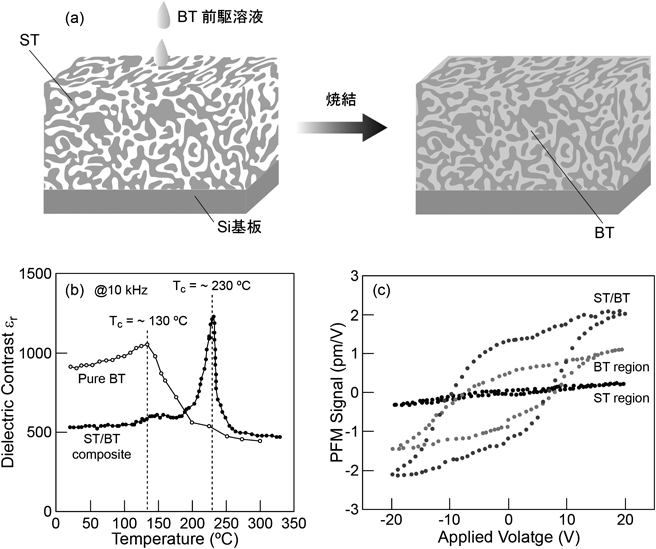
非シリカ系メソポーラス金属酸化物薄膜の新展開Recent Progress of Non-siliceous Mesoporous Metal Oxide Films
1 (独)物質・材料研究機構 若手国際研究センターInternational Center for Young Scientists (ICYS), National Institute for Materials Science (NIMS) ◇ 〒305-0047 茨城県つくば市千現1-2-1
2 (独)物質・材料研究機構 WPI-国際ナノアーキテクトニクス研究拠点World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS) ◇ 〒305-0044 茨城県つくば市並木1-1
受理日:2013年7月30日Accepted: July 30, 2013
発行日:2013年9月30日Published: September 30, 2013