新しい機能の賦与1–3)や新規構造物質の創製4–6)を目的とした,シリケート骨格へのヘテロ金属元素導入は,これまでに数多く行われている。多くの場合,骨格を構成するケイ素原子を同型置換する形で,正四面体型4配位構造をとるヘテロ原子が骨格導入されているが,Engelhardの研究者らにより合成された多孔質チタノシリケートETS-4およびETS-107,8)の場合は,4配位のケイ素,チタン原子とともに,6配位をとるチタン原子が骨格内に導入されているという興味深い構造を有している。このような4配位以外の配位構造をもつヘテロ金属,すなわちヘテロ配位金属を骨格にもつ物質は,International Zeolite Associationからは「ゼオライト」とはみなされない9)。しかし見方を変えれば,だからこそヘテロ配位金属の導入は,「ゼオライト」では実現できない新奇な骨格構造の構築が期待できる有望なアプローチだともいえる。
ヘテロ配位金属種を骨格にもつシリケート物質は決して珍しいものではなく,前述のETS-4, ETS-10以外にもいくつもの物質が報告されている。天然鉱物では,6配位カルシウムを骨格として持つRhodesite(KHCa2Si8O19·5H2O)10)や,同じく6配位のイットリウムを含有するMontregianite(Na4K2Y2Si16O38·10H2O)11,12)などが知られている。RochaらはMontregianiteの同型物質であるAV-113) や,その骨格にユーロピウムやテルビウムのようなランタノイドを導入した物質(Na4K2Ln2Si16O38·xH2O)14,15)の合成について報告している。アルカリ土類金属を含有するシリケートでは,Rhodesiteに類似した結晶構造をもつカルシウム含有シリケートCAS-1(K4Ca4Si16O38·8H2O)16)や,ストロンチウム含有シリケートAMH-3(Na8Sr8Si32O76·16H2O)17)などが合成されており,その構造も解明されている。
我々もカルシウムやストロンチウムなどのアルカリ土類金属をヘテロ配位金属種として用い,それらを骨格導入することにより新規結晶性シリケート物質を合成することを試みた。しかし,ゼオライト様物質の合成に対して一般的に用いられる手法,すなわちシリカ源とヘテロ金属源を含む水性ゲルを水酸化物イオンあるいはフッ化物イオン存在下で水熱合成するという手法を,アルカリ土類金属含有シリケートの合成に適用するには問題がある。このような合成条件下ではアルカリ土類金属は水酸化物あるいはフッ化物の難溶性沈殿を作り,ケイ素との複合物質を形成させるのが難しいのである。それでは前述した既報の合成物ではどのようにこの問題を克服しているかというと,水酸化物がある程度溶解するような比較的高い合成温度で水熱処理することによりケイ素との複合物質を得ているのである。例えばAV-1は230°Cで,AMH-3は200°C,CAS-1は220°Cでそれぞれ水熱合成している。一方,我々はメカノケミカル反応を利用することによりこの問題の克服を試みた。メカノケミカル反応については本誌24号の解説18)を参照していただきたいが,簡単にいえば機械的なエネルギーを加えることにより促進される固相化学反応である。単純で力任せな反応のように見えるが,それだけに様々な固体物質に適用可能であり,簡単な操作で実行可能であることがこの反応の利点である。我々はすでにこのメカノケミカル反応をゼオライト合成に適用し,安価な原料から,簡単な操作で,再現性良くチタノシリケート型ゼオライトを合成できることを報告19–22)している。
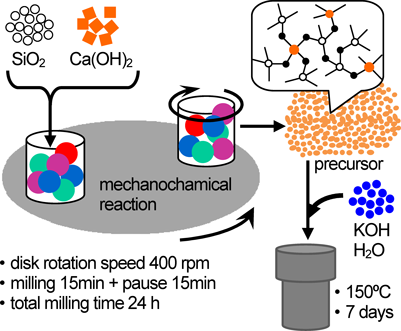
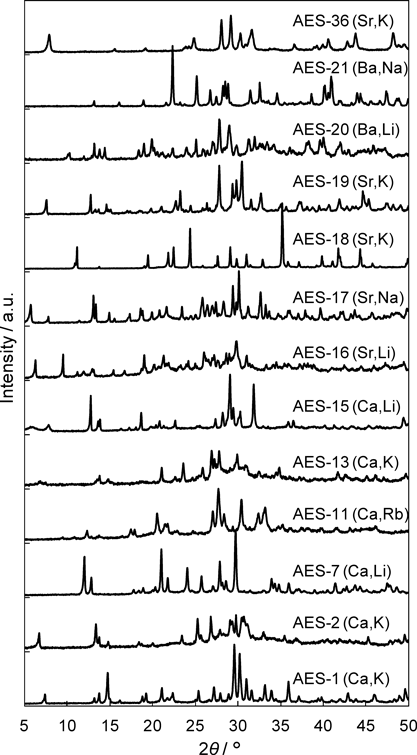
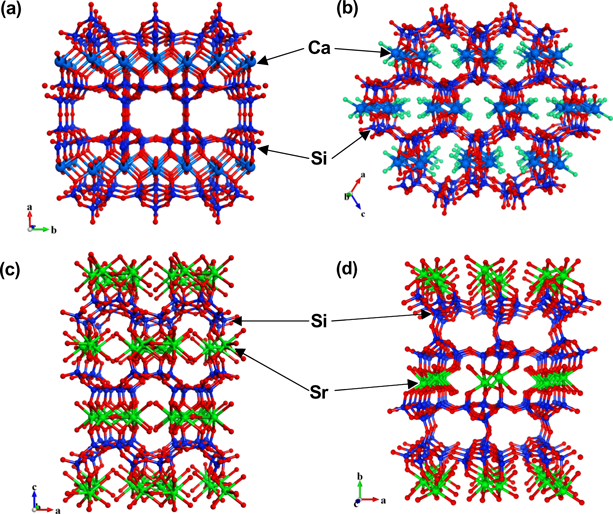
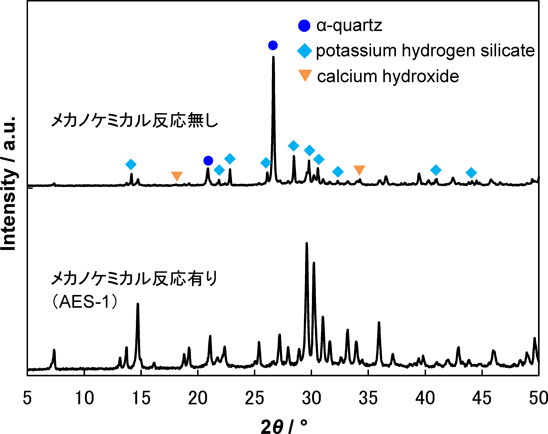
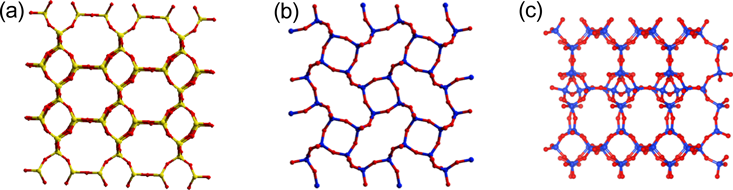
本研究では,アルカリ土類金属水酸化物とヒュームドシリカを出発物質とし,これらと窒化ケイ素製粉砕ボールを,同じく窒化ケイ素製の粉砕容器中に入れ,遊星ボールミルにより高速で粉砕させることにより前駆体を調製した。この前駆体をアルカリ金属水酸化物水溶液中,150°Cで7日間水熱処理して最終生成物を得た(図1)。アルカリ土類金属を含有した前駆体からこれまでに得られた物質の粉末XRDパターンを図2に示す。骨格導入するアルカリ土類金属種や,水熱合成時に加えるアルカリ金属種により生成相が変化し,様々な物質が得られることがわかる。未知物質のうち,粉末X線構造解析により構造が決定された物質の結晶構造図を図3に示す。なお粉末XRDによる構造解析の詳細は文献23を参照されたい。また結晶構造図は可視化プログラムVESTA324)を用いて作成した。
カルシウム含有シリケートAES-125)は前述のCAS-1と同型の物質であり,Ca/Si=0.20~0.30程度のモル比をもつ前駆体を水酸化カリウム水溶液中,150°Cで水熱合成することにより得られる物質である。このAES-1は2次元8員環細孔をもつ多孔性物質であり,多孔性シリケートレイヤーの層間に6配位カルシウム原子が位置している(図3a)。この細孔内に水分子が吸着し,またその吸脱着が可逆的なものであることが確認されたが,より大きな分子径をもつ窒素についてはマイクロ孔への吸着は見られなかった。二酸化炭素分子も細孔内への吸着が見られるが,その吸着等温線を得るには1500秒以上という長い吸着平衡時間が必要であることから,AES-1の細孔径は二酸化炭素の分子径(0.31 nm)と同程度だと考えられる。
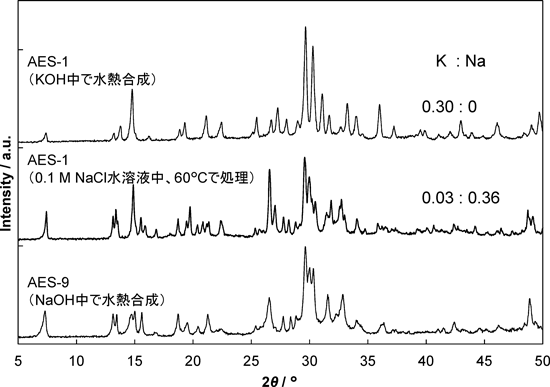
AES-1と同じ前駆体を水酸化ナトリウム水溶液中で水熱処理すると,AES-1とは異なるXRDパターンを示す物質AES-9が得られるが,AES-9とAES-1は同じトポロジーをもち,対称性が異なるために異なるXRDパターンを示すことがわかった(図4)。また,AES-1を塩化ナトリウム水溶液中で処理することによりAES-9が,AES-9を塩化カリウム水溶液中で処理することによりAES-1が得られることがわかっており,KとNaは互いにイオン交換可能であることも見いだされた。このように,2次元酸素8員環をもつAES-1は,水分子を可逆的に吸脱着し,イオン交換可能な骨格外カチオンを有しているという,ゼオライトと同様の物性を有することがわかる。
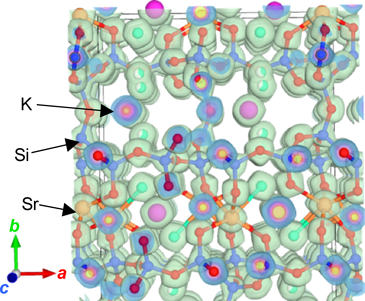
ストロンチウム含有シリケートAES-1926)(図3d)は,カルシウムの代わりにストロンチウムを用いた以外はAES-1と同じ合成条件で得られる物質である。結晶構造解析により,この物質は新規構造物質であり,6配位ストロンチウムがシリケート層を架橋した構造をとることが確かめられた。同じく新規構造物質であるAES-1827)(図3c)がほぼ同じ合成条件で得られるが,AES-18を単一相として得るのは難しく,多くの場合AES-19との混合物として得られ,AES-19のみができてしまうことも多い。なお,AES-18では,7配位のストロンチウムからなるSr4O17のクラスターのレイヤーがあり,それがシリケートレイヤーを架橋した結晶構造をとっている。
AES-19のカリウムはルビジウムとイオン交換可能であるが,ナトリウムとはイオン交換できなかった。興味深いことに,AES-19の前駆体をルビジウム存在下で水熱処理するとAES-19が得られるが,(イオン交換されない)ナトリウムの存在下で水熱処理してもAES-19は得られなかった。ちなみに,ナトリウム存在下で結晶化する物質AES-17は前述のAMH-3と同型の化合物である。AES-19も,AES-1と同様の吸着挙動を見せるが,二酸化炭素分子の吸着挙動から考えると,細孔径はAES-1よりやや大きいと考えられる。
最大エントロピー法(MEM)により得られたAES-19の電子密度分布図を図5に示す。この電子密度分布から結合の共有結合性に関する情報が得られ,共通結合性の強いケイ素原子と酸素原子の間には一様に電子が分布していることがわかる。一方,カリウムの周囲の電子分布は球形をしており,カリウムカチオンが骨格とイオン結合していることが示唆される。ストロンチウムと酸素の結合の場合は,ストロンチウムの電子密度分布に異方性が現れていることから,完全なイオン性ではなく,共有結合とイオン結合の中間の性質をもつと考えられる。このことは,ストロンチウムはカリウムのようにイオン交換できないが,塩基性水溶液での処理により,炭酸ストロンチウムとして析出するという実験事実と矛盾しない。
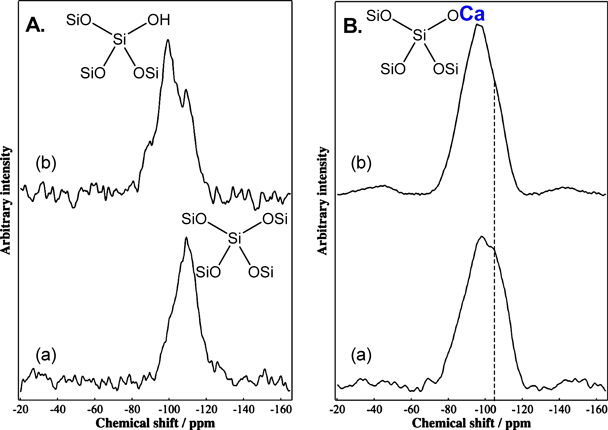
さて,この合成で気になるのが,メカノケミカル反応により前駆体にどのような変化が起こっているのかということである。図6にAES-1の前駆体の29Si MAS NMRスペクトルを示す。メカノケミカル反応前の前駆体のMASスペクトル(図6A. a)ではQ4種に帰属されるピークが−109 ppmに見られる。Q3種はほとんど無いように見えるが,CP/MASスペクトルではSi(OSi)3OHに起因するシグナルが強調され,−99 ppmに大きなピークが見られた。一方,メカノケミカル反応後の前駆体では,MASスペクトル(図6B. a)では,−105 ppmに見えるQ4ピークとともに,−98 ppmにもピークが見られた。しかしこのピークはCPによってそう大きくは強められておらず(図6B. b),CP/MASスペクトルでもQ4ピークがショルダーピークとして見られている。ここから,−98 ppmのピークはSi(OSi)3OHではなく,Si(OSi)3OCaからの寄与が大きいと考えられる。つまり,メカノケミカル反応過程では出発物質それぞれの微粒子化が起こるだけでなく,互いに反応・固溶した物質へと変換されていると考えられる。実際,メカノケミカル反応を経ない出発物質混合物を同じ条件で水熱処理するとケイ酸化合物と水酸化カルシウムの混合物が得られ(図7),ここからも高速粉砕による前駆体調製過程で,物理的な変化だけでなく,化学的な変化が起こったことが示唆される。
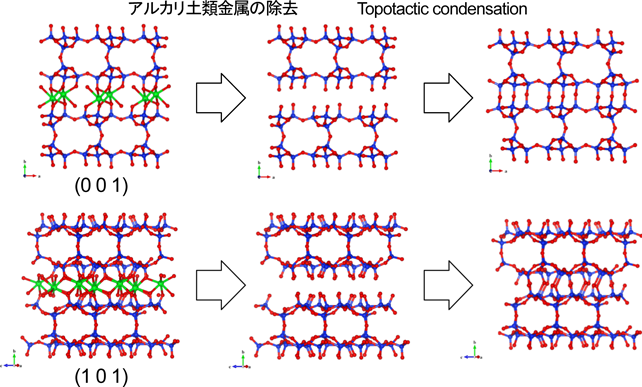
本研究で得られた物質の結晶構造(図3)を見ると,シリケートレイヤーの層間にヘテロ配位アルカリ土類金属原子が位置し,レイヤーを架橋する構造を共通してとっていることがわかる。もしこれらの物質からアルカリ土類金属原子を取り除き,残ったシリケートレイヤーをそのまま縮合させることができれば,これまでにない新たな構造をもつゼオライトを構築することができる。例として,AES-19で想定される構造変換について図8に示す。この場合,ストロンチウムの除去によりc軸方向にも8員環細孔が現れ,3次元8員環ゼオライトへと変換されることになる。本研究で得られた物質には,層を貫く方向に細孔が存在するシリケート層をもつものがあり(図9),このような変換を施したゼオライトあるいは完全剥離によるシリケート層単独での使用が可能となればおもしろい。
このような構造類似性に基づくゼオライトへの構造変換については,Jeongらも彼らが合成したAMH-3に対して提案17)しているが,未だ実現されていない。我々も,弱酸あるいは温水による処理によりストロンチウムが除去できることまでは確認できたが,層状シリケートの構造を保ったまま縮合させることには成功していない。しかし層状シリケートの層間へのゲスト物質の導入・交換や,ゼオライトへの変換についてはすでに多くの研究がなされており,今後の反応条件の詳細な検討により,この新たなゼオライト合成ルートも実現できると期待している。
メカノケミカル反応を利用した,アルカリ土類金属含有シリケート物質の合成手法について簡単に解説した。新しい多孔体合成手法を開発できたこと,そしてその手法を用いることにより様々な未知物質を得ることができたことは大きな成果だと考えているが,未だこの手法の利点を十分に生かした合成ができていないのではないかとも考えている。本手法の重要な利点のひとつは,様々な出発物質に対して,その均質化過程と結晶化過程を明確に分けることができるということである。水溶性の出発物質であれば,その水溶液から均質な水性ゲルを得て前駆体とすることができるが,難溶性の出発物質の場合(例えばETS-10やAV-1のように)水熱処理時に溶解・均質化と結晶化を同条件下で進行させなければならなかった。しかしメカノケミカル反応により固溶させた前駆体を用いれば,溶解条件に縛られることなく,自由に結晶化条件を選択することができる。これにより,細孔容積の大きな多孔性物質の結晶化に有利だと考えられる,より低温での水熱合成が実行可能となるなど,新規多孔性物質の結晶化の可能性を大きく拡張することが可能となる。未だそのような物質合成は実現できていないが,本手法による今後の実現を期待している。
謝辞Acknowledgments
本研究の一部は(財)福岡県産業・科学技術振興財団IST研究FS事業および(独)日本学術振興会科学研究費助成事業(基盤B 24360335)の助成を受けて行われたものである。シリカ源として用いたAerosil200は日本アエロジル株式会社から提供を受けた。本研究中の固体NMRの一部を岐阜大学近江准教授に測定していただいた。この場を借りて深くお礼を申し上げる。
引用文献References
1) M. Taramasso, G. Perego, B. Notari, U.S. Patent, 4,410,501 (1983).
2) A. Corma, L. T. Nemeth, M. Renz, S. Valencia, Nature, 412, 423 (2001).
3) S. H. Park, P. Daniels, H. Gies, Micropor. Mesopor. Mater., 37, 129 (2000).
4) M. J. Annen, M. E. Davis, J. B. Higgins, J. L. Schlenker, Chem. Commun., 1175 (1991).
5) A. K. Cheetham, H. Fjellvåg, T. E. Gier, K. Kongshaug, K. P. Lillerud, G. D. Stucky, Stud. Surf. Sci. Catal. 135, 158 (2001).
6) H. Li, O. M. Yaghi, J. Am. Chem. Soc., 120, 10569 (1998).
7) S. Kuznicki, US Patent, 4, 853, 202 (1989).
8) M. W. Anderson, O. Terasaki, T. Ohsuna, A. Philippou, S. P. MacKay, A. Ferreira, J. Rocha, S. Lidin, Nature, 367, 347 (1994).
9) Ch Baerlocher, L. B. McCusker, D. H. Olson, Atlas of Zeolite Framework Types, 6th revised edition, Elsevier, Amsterdam, p. 371 (2007).
10) E. D. Mountain, Mineral. Mag., 31, 607 (1957).
11) G. Y. Chao, Can. Mineral., 16, 561 (1978).
12) S. Ghose, P.K. Sen Gupta, C. F. Campana, Am. Mineral., 72, 365 (1987).
13) J. Rocha, P. Ferreira, Z. Lin, P. Brandão, A. Ferreira, J.D. Pedrosa de Jesus, J. Phys. Chem. B, 102, 4739 (1998).
14) J. Rocha, P. Ferreira, L. D. Carlos, A. Ferreira, Angew. Chem. Int. Ed., 39, 3276 (2000).
15) D. Ananias, A. Ferreira, J. Rocha, P. Ferreira, J. P. Rainho, C. Morais, L.D. Carlos, J. Am. Chem. Soc., 123, 5735 (2001).
16) J. L. Jordá, S. Prokic, L. B. McCusker, Ch. Baerlocher, C. F. Xue, J. C. Dong, R. Chimie, 8, 331 (2005).
17) K.-K. Jeong, S. Nair, T. Vogt, L. C. Dickinson, M. Tsapatsis, Nature Mater., 2, 53 (2002).
18) 山本勝俊,ゼオライト,24 (2), 43 (2007).
19) K. Yamamoto, S. E. Borjas Garcia, F. Saito, A. Muramatsu, Chem. Lett., 35 (6), 570 (2006).
20) S. E. Borjas Garcia, K. Yamamoto, F. Saito, A. Muramatsu, J. Jpn. Petrol. Inst., 50 (1), 53 (2007).
21) K. Yamamoto, S. E. Borjas Garcia, A. Muramatsu, Micropor. Mesopor. Mater., 101, 90 (2007).
22) S. E. Borjas Garcia, K. Yamamoto, A. Muramatsu, J. Mater. Sci., 43 (7), 2367 (2008).
23) 池田卓史,「粉末X線解析の実際 (第2版)」,中井 泉・泉 富士夫 編著,朝倉書店 (2009),第12章2節,pp. 201–220.
24) K. Momma, F. Izumi, J. Appl. Crystallogr., 44, 1272 (2011).
25) K. Yamamoto, T. Ikeda, C. Ideta, M. Yasuda, Cryst. Growth Des., 12, 1354 (2012).
26) K. Yamamoto, T. Ikeda, C. Ideta, Micropor. Mesopor. Mater., 10.1016/j.micromeso.2013.01.004 (2013).
27) T. Ikeda, C. Ideta, K. Yamamoto, Z. Kristallogr., 10.1524/zkri.2013.1591 (2013).