1.1 ラクトンの環境調和型有機合成法確立をめざして
ラクトンは有用天然化合物である果実香気や抗生物質の基本骨格としての性質を有する化合物である1)。すでに多くの合成例が報告されているが,環境に負荷のかかる鉱酸の使用2)や環境負荷軽減を指向した固体酸使用下の反応における副生成物形成3)など解決すべき点が多い。一方,多孔質アルミノケイ酸塩であるゼオライトは,形状特性を有する細孔内にルイス(L)酸点やブレンステッド(B)酸点を有することから低環境負荷な固体酸触媒として注目されている。また,一般的な溶液下の分子内反応においては,反応種どうしの反応抑制のために高希釈条件とすることから多量の溶媒を必要とするのに対し,ゼオライト包接場が反応種の隔離に有効にはたらくことが期待でき,固体試料を用いた無溶媒反応はもちろんのこと,懸濁溶液条件下の反応にあっても多量の溶媒を必要とせず,資源節約の点からも環境に調和した合成法となりうる。
ここでは,筆者らの研究グループが指向した,ゼオライトナノ包接場を利用した,アルケニルカルボン酸誘導体を反応種とする環境調和型ラクトン合成法についていくつかの例を紹介する。ところで,HSAB理論4)によれば,酸素,ケイ素,およびアルミニウムを構成原子とするゼオライトは硬い試薬に分類される。一般的に柔らかい反応に分類される有機反応に対し,硬いゼオライトが有効にはたらく理由を解明できれば,有機反応に対するゼオライトの利用拡大が期待できる。半経験的分子軌道法を用いた解析手順と併せて,解析例の一端を紹介する。
1.2 ゼオライトの触媒作用と固体酸性の発現
ゼオライトの触媒作用と固体酸性の発現についてはすでに多くの成書5–7)で触れられているが,本反応系を理解するうえで必要となるため,その概略に触れることにする。
ゼオライトの触媒特性は,その多様な構造と性質にある。しかも,多くの酸触媒とは異なり,結晶性であるゼオライトは,必要とされる性質を持った物質の選択とその制御が容易である。ゼオライトの触媒作用には,以下に示した多様な性質がいろいろな形で関与する。
- 1)ブレンステッド酸としての作用と酸強度の多様性
- 2)骨格酸素アニオンに基づく塩基性
- 3)骨格外金属イオンおよび遷移金属錯体による触媒作用
- 4)骨格内金属イオンに基づく触媒作用
- 5)細孔構造に基づく選択性の制御(形状選択性)
- 6)細孔内の電場勾配による反応物の分極
- 7)異常電子価,極性中間体の安定化
固体酸性を示す水酸基は,ゼオライト骨格の部分構造に起因しており,次のように導入される。HYゼオライトは,NH4Yを350°C以上で焼成することで得られる。このとき酸性プロトンは,ゼオライト中ではアルミニウムとケイ素に架橋した酸素に結合し,水酸基として存在する。したがって,室温で測定した赤外吸収(IR)スペクトルには水酸基の伸縮振動が観測される。この吸収は,ピリジンの吸着により消失し,代わりにピリジニウムイオンの吸収が出現する。このことは,アルミニウムとケイ素に架橋した水酸基が塩基分子であるピリジンに対してプロトン供与能を有することを示している。すなわち,架橋水酸基はB酸点となる。
2. ゼオライトナノ細孔を反応場とする環境調和型ラクトン合成法の検討8)
2.1 ゼオライトに対する前処理ならびに機器測定
反応に対するゼオライト細孔形状の影響を検討すべく,HMOR, HY, USY, H-ZSM-5などを選択した。ここでは,FAU型ゼオライトであるHYを例として,ゼオライトを活性化するための前処理や機器測定について以下に記す。
HYに対する熱重量分析および示差熱分析(TG/DTA)測定により,脱水に伴う質量減少などを確認した後,500°Cでの長時間維持において質量ならびに熱量に変化が見られなかったことから,反応前の焼成条件を,電気炉による空気下500°C15時間焼成とした。焼成温度の違いによりゼオライトの構造に変化が生じるというLutzの指摘9)を踏まえ,500°Cで焼成したHYに対する粉末X線回折(XRD)を行い,結晶構造の維持を確認した。焼成後のHYを,200°Cまで放冷した後,使用直前までデシケーター中で真空保存した。
岡本らは,XPSよるA, X, Y, MOR型ゼオライトに対するキャラクタリゼーションの結果,外表面組成と内表面組成におけるSi/Al比に関する情報として,未処理のゼオライトではほぼ等しく,アンモニウムイオンでイオン交換後,脱アミノ化により調製したH型では外表面の脱アルミ化が顕著であると報告している10)。そこで,固体NMR測定法11)を用いて,29Si NMR測定によるHY のSi/Al比ならびに SiO4周りのAl置換数の算出,27Al NMR測定による骨格内外のアルミニウムの存否確認を行った。29Si-NMR 測定によるSi/Al比の誘導式を次に示す。
焼成後のSi/Al比とAl置換数とを別個に定量化できることは,本反応系における反応場の指標を入手するうえで極めて重要な意味をもつ。すなわち,Si/Al比がAl置換数のばらつきを平均化した統計的な数値にとどまるのに対し,Al置換数は,その数値の0から3までの分布に基づいた酸強度の均質性に関する定量的な指標となり,反応の最適条件の探索ならびに再現性維持のために極めて重要な役割を果たす。
本実験で用いた合成機器ならびに分析機器は以下のとおりである。
合成機器;電気炉:必要に応じてISUZU Muffle Furnace DSTR-13K, ISUZU Muffle Furnace ATR-11K, AS ONE小型プログラミング電気炉MMF-1のいずれかを使用。
分析機器;TG/DTA測定:DTG-60,溶液試料および固体試料に対するNMR測定:付属設備の交換を伴うJEOL NM-ECA500,IR測定:HITACHI 270-50 Infrared Spectrophotometer,GC-MS 測定:SHIMADZU GC-17Aを使用。
2.2 包接試料の作成
2.2.1 懸濁溶液法
所定量の反応種を加えたシクロヘキサン溶液20 mLを暗所・窒素雰囲気下で30分間撹拌した後,あらかじめ焼成しておいた各種酸性ゼオライトの所定量を加えて再度30分間暗所,窒素雰囲気下で撹拌した。この懸濁液をろ過し,包接試料を作成した。また,ろ別したシクロヘキサン溶液に対する1H NMR 測定を行い,未包接反応種に対する定量結果を用いて包接量を確認した。
2.2.2 ミリング法
アミドなど反応種が固体である場合,懸濁溶液下における包接作業を経由せず,反応種とゼオライトと直接ミリングして試料調製を行うこともできた。
2.3 包接反応および後処理
包接反応は,固相熱浴反応とした。すなわち,所定の温度および時間制御下,電気炉内において包接試料を加熱した無溶媒反応である。一般的な溶液下の分子内反応において必要とする多量の溶媒を全く使用しない点を,強調したい。また,このような固体試料は,熱浴条件に替わるマイクロウェーブ照射実験の適用も容易となる。
反応終了後,包接試料に対するアセトンもしくはジクロロメタン抽出,減圧濃縮を経て,NMRならびにGC-MS測定により生成物の同定・定量を行った。
2.4 実験結果および考察
2.4.1 直鎖アルケニルカルボン酸を反応種とする包接反応―ラクトン化における位置選択性の検討
直鎖カルボン酸の包接反応の結果を表1に示した。3-および4-ペンテン酸を反応種とする100°Cの反応(Entry 1, 2)では,同一生成物として,五員環を有するγ-バレロラクトン(5-methyldihydrofuran-2(3H)-one)のみを高収率で与えた。このような結果に対する機構として,3-ペンテン酸ではendo型閉環が,4-ペンテン酸ではexo型閉環がそれぞれ選択的に進行したとするか,あるいはアルケン部位の転位後に両者とも同一型の閉環が後続したとするかについては,今後の検討を要する。
表1 直鎖アルケニルカルボン酸およびラクトンを反応種とする包接反応1)の結果| Entry | Substrates | Products | Conditions |
|---|
| 100°C 10 h | 60°C 10 h |
|---|
| R= | n= | (R'=) | δ(R''=) | Yield[%] | γ:δ | Yield[%] | γ:δ |
|---|
| 1 | Me | 1 | Me | — | 85 | — | No data | No data |
| 2 | H | 2 | Me | — | 84 | — | 69 | — |
| 3 | H | 3 | Et | Me | 59 | 89:11 | 45 | 29:71 |
| 4 | n-Pr | 1 | n-Pr | Et | 68 | 94:6 | 32 | 96:4 |
| 5 | Et | 2 | n-Pr | Et | 68 | 94:6 | 46 | 56:44 |
| 6 | H | 4 | n-Pr | Et | 57 | 85:15 | 58 | 12:88 |
| 7 | n-Pent | 2 | n-Hex | n-Pent | 25 | 95:5 | 32 | 36:64 |
| 8 | γ-Lactone (R'=Et) | Et | Me | 83 | 93:7 | No data | No data |
| 9 | δ-Lactone(R''=Me) | Et | Me | 58 | 90:10 | No data | No data |
| 1)典型的な反応条件;反応種;100 mg,HY(Si/Al比:3.69); 2.0 g. |
分子鎖長を一つ増やした5-ヘキセン酸の反応(Entry 3)では,五員環ラクトン(5-ethyldihydrofuran-2(3H)-one)と六員環ラクトン(6-methyltetrahydro-2H-pyran-2-one)とが競争的に生成した。興味深いことにこれらの生成比には温度依存性が観測された。高温における優先生成物である五員環ラクトンならびに低温における優先生成物である六員環ラクトンは,それぞれ熱力学的支配,速度論的支配によるものと考察される。ここで生成した五員環ならびに六員環ラクトンを新たな反応種として,それぞれの包接反応を100°C下で検討した結果(Entry 8, 9),ラクトンの生成比は,5-ヘキセン酸の反応結果(Entry 3)とほぼ等しい9:1の比を与えた。このことは,包接反応中に両ラクトン間の平衡過程が存在する可能性を示唆した。
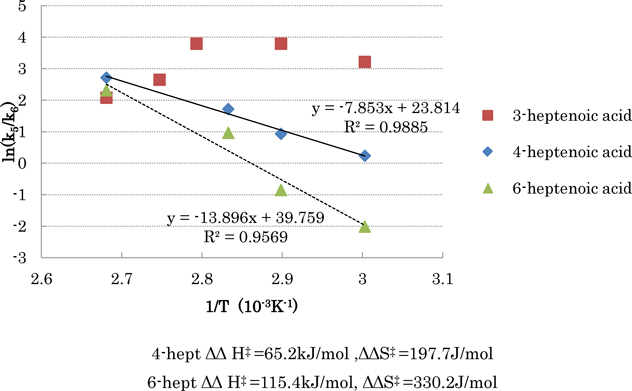
さらに鎖長を一つ増やした3-, 4-および6-へプテン酸の反応(Entry 4, 5, 6)においても五員環ラクトン(5-n-propyldihydrofuran-2(3H)-one)と六員環ラクトン(6-ethyl tetrahydro-2H-pyran-2-one)とが競争的に生成した。3-へプテン酸における六員環ラクトン生成については,アルケン部位の転位を経由する過程,もしくはアルケン部位の転位を必要とせずに生成できる五員環ラクトン生成後の平衡を経由する過程が考えられる。これに対して,6-へプテン酸からの五員環および六員環ラクトンの生成は,アルケン部位の転位過程無くしては生成できない。3種のへプテン酸の反応に対する温度効果の詳細な検討については,2.4.2で述べる。
4-デセン酸の反応(Entry 7)から得られた五員環および六員環ラクトン(それぞれ,5-n-hexyldihydrofuran-2(3H)-one, 6-n-pentyltetrahydro-2H-pyran-2-one)は,いずれもモモの香料成分として知られる化合物1)であり,有用天然物の環境調和型合成の一例とすることができた。また,それらの位置選択性は,5-ヘキセン酸の反応(Entry 3)に類似する温度依存性を示した。
2.4.2 3-, 4-および6-へプテン酸の反応における位置選択性に対する温度効果の検討
検討結果をEyring plotとして図1にまとめた。五員環ラクトンと六員環ラクトンの生成が同じ中間体を経由すると仮定し,絶対温度の逆数に対する両ラクトン生成比の自然対数値をEyring plotした場合,同一生成物を与える3種の反応種に対して,全く同一の直線が得られるはずである。ところが,3-へプテン酸の反応では曲線を,4-および6-へプテン酸の反応では傾きの異なる直線を与えた。この結果は,へプテン酸の競争的ラクトン化の律速段階として複数の因子が関与していることを示唆する。たとえば,分子鎖の内側から外側へのオレフィン部位の転位,分子鎖の外側から内側へのオレフィン部位の転位,さらにラクトンに至る閉環過程などがその候補となりうる。今後,さらに詳細な反応機構の解析を必要とする。
2.4.3 5-ヘキセン酸を反応種とする再利用ゼオライトによる包接反応
環境調和型合成法としての観点から,一度反応に使用したゼオライトを再度焼成することで再利用することができれば,資源の節約上たいへん好ましい。5-ヘキセン酸を用いた検討結果を表2に示した。初回から4回目へ推移するに従い,生成量の若干の低下とともに,六員環ラクトンに対する五員環ラクトンの優先性が低下した。
表2 5-ヘキセン酸を反応種とする再利用ゼオライトによる包接反応の結果| 利用回数 | HY1) [g] | Sub. [mg] | Sub./S.C.2) | Temp.[°C] | Time [h] | Products [%] |
|---|
| Total yield | Recov. | Lactone | Others3) | Ratio |
|---|
| γ | δ | γ:δ |
|---|
| 1 | 45.6 | 1.1 | 1/2.0 | 100 | 10 | 69 | n.d. | 57 | 8 | 4 | 89:11 |
| 2 | 51.7 | 1.1 | 1/1.8 | 100 | 10 | 62 | 0.6 | 34 | 23 | 5 | 60:40 |
| 3 | 52.9 | 0.9 | 1/1.3 | 100 | 10 | 69 | n.d. | 32 | 32 | 5 | 49:51 |
| 4 | 41.3 | 0.7 | 1/1.4 | 100 | 10 | 51 | 0.8 | 23 | 23 | 5 | 50 : 50 |
| 1)再生条件;すべて500°C,15時間に統一.XRDの測定結果;すべてにおいてアモルファス化は確認されず.29Si NMR の測定結果;初回および4回目のSi/Al比は54±2%でほぼ一定.27Al NMR の測定結果;[骨格内4配位:骨格外5配位:骨格外6配位]の値が,初回の[17.2:69.3:13.5]から4回目の[12.8: 68.2:19.0]に変化した.2)ゼオライトのスーパーケージに対する反応種の比.3)アルケンの転位に伴う3-および4-ヘキセン酸の生成. |
小野らは,ゼオライトの固体酸性はゼオライト骨格を形成するSi/Al比の影響を受け,Si/Al比の上昇は以下のような特性を示すことを指摘している7)。
1)固体酸密度が低下し,水素移行反応が抑制される。
2)結晶の安定性が向上し,耐水熱性が高くなる。
3)静電場強度が減少するため,疎水性分子を吸着しやすくなる。
1)の水素移行反応の抑制は,ラクトン形成の位置選択性に対しても大きな影響を与える。表2の脚注に示したとおり,焼成を繰り返したゼオライトに対する種々の測定の結果,XRDにおいては構造のアモルファス化は確認されなかった。29Si NMRではSi/Al比に変動は見られなかったものの,27Al NMRで骨格内4配位Alの減少および骨格外6配位Alの増加に伴うゼオライト内表面のSi/Al比の上昇が確認された。これは岡本らが報告した焼成による外表面への脱Al化と一致する10)。ゼオライト内表面のAlの脱離は,計算においてB酸点のLUMOのエネルギー準位の上昇をもたらし,かつB酸点の減少により熱力学的支配条件で生成する五員環ラクトンの生成を抑制する結果となった。したがって,ゼオライトの再利用に際して反応の再現性を確保するためには,固体NMR測定による内表面のSi/Al比の確認が必要であることが明らかとなった。次節では,さらに理解を深めるためにSi/Al比の異なるHYを用いた包接反応を検討することにした。
2.4.4 4-ペンテン酸を反応種とするSi/Al比の異なるHYを用いた包接反応
2.1で述べたとおり,焼成後のSi/Al比とSiO4周りのAl置換数とを別個に定量化できることは,本反応系における反応場の指標を入手するうえで極めて重要な意味をもつ。4-ペンテン酸を用いた検討結果を表3に示した。表中には,焼成後の各HYに対するAl置換数の分布も掲載した。懸濁下,室温30分の反応にも関わらず,最も高い46%の収率で五員環ラクトンを生成したHSZ-331HSAにおけるSi/Al比が11.1, Si(1Al)の比率が36.0%であった(Entry2)。これらの数値は,本反応系に対する最適条件として,3で述べる分子軌道論を用いた反応場構築の際の参照値として活用することにした。
表3 4-ペンテン酸を反応種とするSi/Al比の異なるHYを用いた包接反応1)の結果| Entry | HY zeolite | Si/Al比2) | Al置換数の分布[%]2) | Yield [%]3) |
|---|
| 0 | 1 | 2 | 3 | Recover | Product |
|---|
| 1 | CBV-600 | 55.8 | 92.8 | 7.2 | — | — | 61.7±8.6 | 10.0±3.6 |
| 2 | HSZ-331HSA | 11.1 | 64.0 | 36.0 | — | — | 25.8±2.7 | 46.3±2.2 |
| 3 | JRC-Z-HY5.5 | 4.68 | 39.9 | 34.6 | 25.5 | — | 74.8±2.8 | 2.6±0.7 |
| 4 | Lot.No.101217 | 2.83 | 22.3 | 31.2 | 29.3 | 17.2 | 61.4±5.1 | 3.4±0.2 |
| 1)典型的な反応条件;反応種;50 mg,HY(Si/Al比:3.69); 1.0 g,シクロヘキサン懸濁溶液条件下,室温,30分撹拌.2)煆焼後の各HYに対する 29Si NMR 測定による値.3)生成物;γ-バレロラクトン(5-methyldihydrofuran-2(3H)-one). |
2.4.5 側鎖を有するアルケニルカルボン酸を反応種とする包接反応―ラクトン化における立体選択性の検討
4-ペンテン酸の3位にメチル基もしくはフェニル基を導入したラセミ体の反応種を用いた検討結果を表4に示した。両反応種とも,五員環ラクトンの生成率は,母骨格の等しい4-ペンテン酸の反応結果(表1, Entry 2; 100°C下の収率;84%)と同等の値を示し,それぞれ80%,78%となった。興味深いことは,各々の立体選択性が逆の温度依存性を示したことである。すなわち,メチル置換体では,高温時におけるcis体の優先生成(熱力学的支配)ならびに低温時におけるtrans体の優先生成(速度論的支配)を示したのに対し,フェニル置換体では,高温時におけるtrans体の優先生成(熱力学的支配)ならびに低温時におけるtrans体生成率の減少(速度論的支配下におけるcis体の増加)が観測された。このような立体選択性に対する制御要因の解明は,多くの天然物にみられる光学異性体の生成機構に関連する重要な課題であり,継続的な検討を必要とする。
表4 側鎖を有するアルケニルカルボン酸を反応種とする包接反応1)の結果| Entry | Substrates | Products | Conditions |
|---|
| 100°C 10 h | 60°C 10 h |
|---|
| cis | trans | Yield[%] | C:T | Yield[%] | C:T |
|---|
| 1 |  |  | 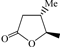 | 80 | 55:45 | 78 | 41:59 |
| 2 | 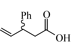 | 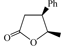 | 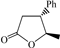 | 78 | 30:70 | 51 | 41:59 |
| 1)典型的な反応条件;反応種;50 mg, HY(Si/Al比:3.69); 1.0 g. |
2.4.6. 環状部位を有するアルケニルカルボン酸を反応種とする包接反応
環状部位を有する2-シクロペンテン-1-酢酸および1-シクロヘキセン-1-酢酸を反応種とした検討結果を表5に示した。前者の100°Cの反応では,穏やかな収率で単一生成物である五員環ラクトン(2-oxabicyclo [3, 3, 0] octan-3-one)を与えたのに対し,60°Cの反応では,五員環ラクトンを主生成物とする六員環ラクトン(2-oxabicyclo[3, 2, 1]octan-3-one)の競争的生成が観測された(Entry 1)。後者の100°Cおよび60°Cの反応では,痕跡量の五員環ラクトン(7-oxabicyclo [4,3,0] nonan-8-one)の生成を確認したが定量には至らなかった(Entry 2)。環状アルケン部位の立体的制約がラクトン化に大きな影響を及ぼしたと考えられるが,このような環状部位を有するカルボン酸の反応性を明らかにすることは,ワインラクトンなど多環性天然化合物を合成する上で重要な知見となることから,継続的な検討を進めていきたい。
表5 環状部位を有するアルケニルカルボン酸を反応種とする包接反応1)の結果| Entry | Substrates | Products | Conditions |
|---|
| 100°C 10 h | 60°C 10 h |
|---|
| γ-Lactone | δ-Lactone | Yield[%] | γ:δ | Yield[%] | γ:δ |
|---|
| 1 |  |  |  | 58 | 100:0 | 54 | 84:14 |
| 2 |  |  | — | trace | — | trace | — |
| 1)反応種;50 mg, CBV-600(Si/Al比:55.8); 1.0 g.2)Trace; NMRにより生成物の確認はできたが,反応種のピークと重なり定量できなかった. |
2.4.7 エステル,ニトリル,ならびにアミドを反応種とする包接反応
標題に示した4種の4-ペンテン酸誘導体を反応種とした包接反応の結果を表6-1に,4種の5-ヘキセン酸誘導体を反応種とした包接反応の結果を表6-2に示した。
表6-1 4-ペンテン酸,エステル,ニトリル,ならびにアミドを反応種とする100°Cでの包接反応の結果| Entry | Substrates | HY [g] | Sub./S.C. | Time (h) | Products [%} |
|---|
| n=2/X | [mg] | Total | Recov. | Lactone | Others | Ratio |
|---|
| γ (R'=Me) | δ (R''=H) | γ:δ |
|---|
| 1 | -COOH | 91.6 | 2.0 | 1/2.0 | 10 | 84 | n.d. | 84 | — | n.d. | — |
| 2 | -COOEt | 48.2 | 1.2 | 1/2.2 | 1 | 68 | n.d. | 68 | — | n.d. | — |
| 3 | -CN | 100.8 | 1.9 | 1/1.0 | 24 | 49 | 23 | 16 | — | 101) | — |
| 4 | -CONH2 | 44.7 | 1.0 | 1/1.5 | 20 | 48 | 17 | 31 | — | n.d. | — |
| 1)アミド形成. |
表6-2 5-ヘキセン酸,エステル,ニトリル,ならびにアミドを反応種とする100°Cでの包接反応の結果| Entry | Substrates | HY [g] | Sub./S.C. | Time (h) | Products [%} |
|---|
| n=3/X | [mg] | Total | Recov. | Lactone | Others | Ratio |
|---|
| γ (R'=Et) | δ (R''=Me) | γ:δ |
|---|
| 1 | -COOH | 89.9 | 2.0 | 1/2.0 | 10 | 58 | n.d. | 52 | 6 | n.d. | 89:11 |
| 2 | -COOEt | 45.7 | 1.1 | 1/1.8 | 10 | 56 | n.d. | 50 | 6 | n.d. | 89:11 |
| 3 | -CN | 46.8 | 1.0 | 1/1.4 | 15 | 6 | 2 | 1 | 3 | n.d. | 25:75 |
| 4 | -CONH2 | 45.7 | 1.0 | 1/1.4 | 10 | 47 | 36 | 1 | 10 | n.d. | 9 : 91 |
4-ペンテン酸誘導体の反応では,主生成物として同一の五員環ラクトンを与えたが,生成率の低いニトリルおよびアミドの反応に比べて,カルボン酸およびエステルからの収率は良好な値を示した。4-ペンテンニトリルの反応では,副生成物として4-ペンテンアミドが観察された。
5-ヘキセン酸誘導体の反応では,五員環および六員環ラクトンの競争的生成が観察された。エステルはカルボン酸と同等の反応性を示した。これに対して,ニトリルおよびアミドの反応は,カルボン酸やエステルと比較して低く,ラクトン化の位置選択性が逆転する結果を与えた。
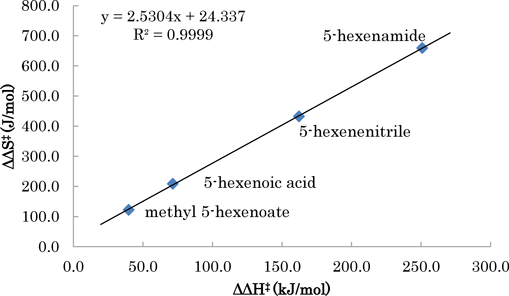
5-ヘキセン酸をはじめとする4種の誘導体を反応種とした包接反応における五員環および六員環ラクトンの競争的生成に対する温度効果を比較検証した。各反応種に対して得られた活性化パラメータを表7に示した。これらの値を用いて作成したIsokinetic plotを図2に示す。図2が示す4種の反応種に対する極めてよい直線性は,これら4種の競争的反応が同一の反応機構を経由することを示唆する。
表7 5-ヘキセン酸,エステル,ニトリル,ならびにアミドを反応種とした五員環および六員環ラクトンの競争的生成に対する活性化パラメータ| Substrates | ΔΔH‡ (kJ/mol) | ΔΔS‡ (J/mol) |
|---|
| methyl 5-hexenoate | 39.7 | 122.5 |
| 5-hexenoic acid | 71.5 | 209.0 |
| 5-hexenenitrile | 162.2 | 432.6 |
| 5-hexenamide | 250.8 | 659.6 |
当初,ニトリルおよびアミドを反応種とした実験は,ラクタムの生成を期待したが,いずれの場合もラクトンを主生成物として与えた。焼成したゼオライトを用いたにも関わらず,ニトリルの反応が含酸素化合物のラクトンを与えたことは,反応系中に水が残存したことを示唆する。結果として,ゼオライト包接場を用いたラクトン合成における反応種としての適性範囲が,カルボン酸やエステルにとどまらずニトリルやアミドも許容であることを明らかにした。
3.1 ゼオライト内表面に関する実験による定量的解析
岡本らは,XPSよるA, X, Y MOR型ゼオライトに対するキャラクタリゼーションの結果,2.1.1で触れたSi/Al比に関する情報に加えて,次のような指摘を行っている10)。ゼオライトに対して吸着・触媒能を期待する際,細孔内のカチオン分布,カチオンや格子酸素の電子状態に関する知見が重要であり,O1s準位の結合エネルギー(BE)は,Si/Al組成比に依存する。このBEの変化を左右する格子酸素の電荷密度は,ほとんど構造に依然せずアルミニウム含有量と共に増加する。XPSで求めたO1s準位のBEがゼオライト格子酸素の塩基強度の良い尺度となる。また,ゼオライト細孔内の塩基サイトの不均質性についても言及している。一方,ゼオライト中のルイス(L),ブレンステッド(B)各酸点のXPSによる定量化は,各種ゼオライトに対するピリジン吸着の測定により行われ,H-ZSM-ピリジン系に対する測定値;398.7, 400.3, 402.1 eVが,それぞれL酸点,弱いB酸点,強いB酸点に帰属,定量化されている。
3.2 ゼオライトを包接場とした有機反応に対する分子軌道論的解析
すでに,メタノールのオレフィン化(MTO)反応12)や,アルケンの転位反応13)に対する非経験的分子軌道計算の適用例が報告されている。ゼオライトに対する扱いは,前者が最小鎖状Si–O–Al–O–Siクラスター構造を,後者は環状構造を含む54Tクラスター構造を採用している。一方,MOPAC14)に代表される半経験的分子軌道法の適用は,バージョン更新毎にパラメータの充実が図られているにもかかわらず,ゼオライト系への適用はふさわしくないといわれている。
環境調和型有機合成を志向した筆者らの分子内反応場の設計に際し,計算化学が評価すべき事項は,以下のとおりである。
- 1)包接前の溶液下,配座平衡状態にある反応種の配座異性体に対する包接選択性(特に,閉環に必要な配座異性体に対する選択性)
- 2)包接後の反応種における配座平衡の存否
- 3)包接前後の反応種配座平衡における相違の有無
- 4)目的とする分子内反応の包接下における進行の成否
- 5)反応種(または目的生成種)の包接に伴う安定化エネルギーの相違確認
包接の可否については,反応種・包接場おのおのに対する分子模型の比較から容易に判断できると思われがちだが,サイズ的には包接可能であっても,安定化が伴わない場合は困難となることが予測される。さらに,目的とする反応の成否を予見するためには,上述した1)から4)の各項目に関する定量的な検討が必要となる。すでにXPSによる実験解析でその重要性が指摘されているとおり,ゼオライト内表面のL, B各酸点および塩基点のエネルギー準位およびそれらの分布の確認が不可欠であり,各サイトと反応種との相互作用に伴う動的過程の解析が必要となる。5)については,目的生成種が,後続反応種と自発的に入れ替わる可能性を予見することにより,抽出操作の有無など,反応後の処理法の選択上,極めて重要な知見となる。
以上の観点から,本系に対する鎖状クラスターモデルの適用には限界があり,包接内表面部位の最小単位と反応種との間の動的過程の評価が必要となる。筆者らの研究室では,計算化学におけるソフト,ハードを含めた経済的コストならびに計算時間コストの観点から,非経験的分子軌道法の適用が困難であり,改めて半経験的分子軌道法の利用を図ることとし,併せてゼオライト系への適用が適切ではないとされた原因を探ることにした。
3.3 半経験的分子軌道法の適用に向けて
3.3.1 PM5, PM6およびPM7法による検証
ゼオライトの最小単位構造に相当するテトラヒドロキシシランやジシロキサン15)に対する構造最適化をPM5, PM6およびPM7の各法を用いて行った結果,ケイ素–酸素–水素(もしくはケイ素)の構造に対して,PM5法では曲線構造を与えたのに対し,PM6およびPM7法では直線構造を与えた。これにより,ゼオライトの基本骨格であるケイ素-酸素-ケイ素構造に対するPM6およびPM7法の適用が問題視される理由の一端が明らかになった。この結果を踏まえ,以後の計算にはPM5法を用いることとした。
3.3.2 水素原子によるキャッピングの影響について
計算化学におけるゼオライトの部分構造切り出しの際,ケイ素およびアルミニウム原子における骨格形成の残余価標に対する扱いとして,構造の自由度が増える水酸基に替えて水素原子を用いる場合が多い。そこで,テトラヒドロキシシランの水酸基を順次水素原子で置換した化合物の最適化を行い,おのおのの標準自由エネルギー(ΔH0),最高占有軌道(HOMO)エネルギー準位,最低非占有軌道(LUMO)エネルギー準位の各数値に関する変遷を比較検討した。それらの結果を表8にまとめた。切り出した骨格に対する計算を行う際のケイ素およびアルミニウムの残余価標は1ないし2となることから,一ないし二水素置換体とテトラヒドロキシシランとのΔH0, HOMO, LUMOの数値の格差には留意を要する。たとえば,n=0と2との間の各数値の格差は,それぞれ723.8 kJ/mol, 0.95 eV, 0.61 eV となった。
表8 SiOH(4−n)Hnに対する標準自由エネルギー(ΔH0),最高占有軌道(HOMO)エネルギー準位,最低非占有軌道(LUMO)エネルギー準位の変遷1)| Hn | ΔHo [kJ/mol] | Δ [kJ/mol] | HOMO [eV] | Δ [eV] | LUMO [eV] | Δ [eV] |
|---|
| 0 | −1312.4 | 0.0 | −11.024 | 0.00 | −0.697 | 0.00 |
| 1 | −957.7 | 354.7 | −10.68 | 0.34 | −0.227 | 0.47 |
| 2 | −588.6 | 723.8 | −10.07 | 0.95 | −0.0892 | 0.61 |
| 3 | −243.6 | 1068.8 | −9.93 | 1.09 | 0.1498 | 0.85 |
| 4 | 44.0 | 1356.4 | −11.22 | −0.20 | 1.29 | 1.99 |
| 1)FUJITSUのライセンスを取得したMOPAC2002 ver.3.5/PM5法による. |
3.3.3 ゼオライト包接モデルの構築と評価
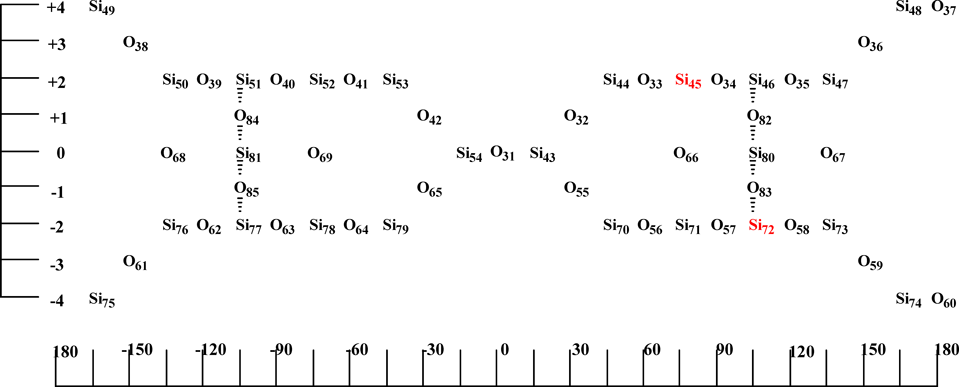
PM5法を用いた反応種-ゼオライト包接系構築のための作成手順を,対称性の扱いが容易なH-モルナイトと4-ペンテン酸との系を例として,以下に示す。H-モルデナイトの8員環部と12員環部を同時に含み,C2v対称を有する部分構造(Si=22, O=28)の展開図を図3に示した。この展開図を参考にPM5法適用のための入力データを作成した。作成上の留意点は,以下のとおりである。
ゼオライト主骨格の入力は,構成原子間の結合情報を全く採用せず,カルテシアン座標の原点を含むxy平面を基準としてz軸方向に一定の間隔をおいた上下各4面からなる9枚の各面上に対称性を配慮して配置した。この際,各基準面の(x, y=0, 0)点と各酸素原子との距離設定として,細孔の短軸および長軸に関する文献値を参考とした以外は,任意の値を用いた。この入力法は,カルテシアン座標適用下にあっても,入力順序の後位にある原子の最適化が前位原子の構造パラメータ変動の影響を受けないため,最適化に要する計算時間が短縮できる。また,キャッピング水素の入力は,sp3混成ケイ素原子との結合を想定した結合長,結合角,ならびに二面角を採用した。以下に段階的構造最適化の手順を示す。
1)全原子のパラメータを固定し,対称的な上下各4面の間隔のみを最適化した。
2)上記操作後の基準面9枚を固定した後,対称性を踏まえつつキャッピング水素に対して結合長,結合角,ならびに二面角を最適化した。
3)上記操作後のキャッピング水素を固定した後,主要原子の二面角に相当するパラメータを対称性に配慮しながら最適化した。この操作は,1)の最適化に際して各基準面上にあった主要原子を,基準面の上下方向に,より精密に最適化することに相当する。
4)上記操作の結果を固定した後,各基準面の(x, y=0, 0)点と主要原子間の距離および角度を,対称性に配慮しながら最適化した。なお,細孔の短軸および長軸に相当する数値を入力値のままとすることもできる。
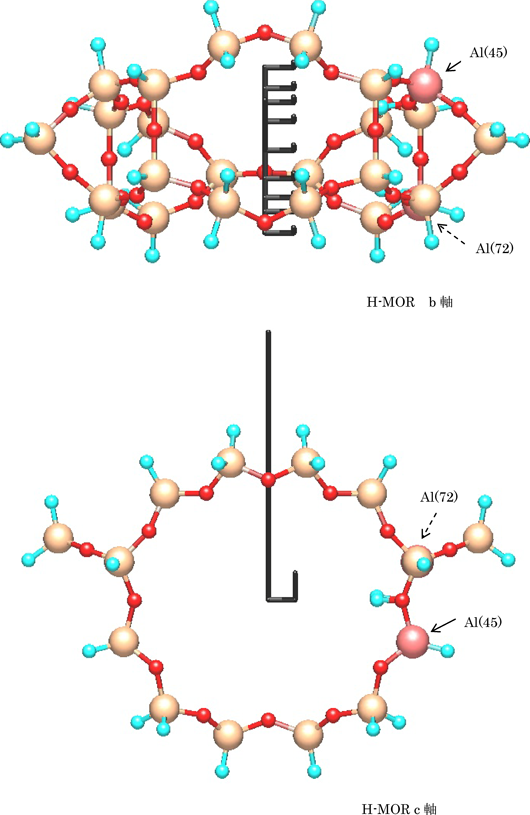
5)上記操作の結果を固定した後,本反応の実験により得られた(2.4.4および表3参照)適切なSi/Al比(≒11.1)ならびにTO4四面体におけるアルミニウム原子置換数(=1)を参考に決定したアルミニウム2原子を,Lowenstein 則(Al–O–Al 回避)7),五員環2(Al)回避則7),Dempsey則(六員環に対するメタ置換優先)7)に従って導入した(図3のSi(45)およびSi(72)を置換)。併せて,この置換に伴って生じる2つの負電荷を補償すべく2つの水素イオンを補填(表9,ダミー原子XXから,位置可変として置換された水素原子(86)および(87)に相当)した。
この操作終了後に得たH-モルデナイトのカルテシアン座標ならびに構造図を,それぞれ表9,図4に示した。
表9 展開図3を出発点とした,3.3.3,操作5)段階のAl-2置換H-MOR骨格のカルテシアン座標データベース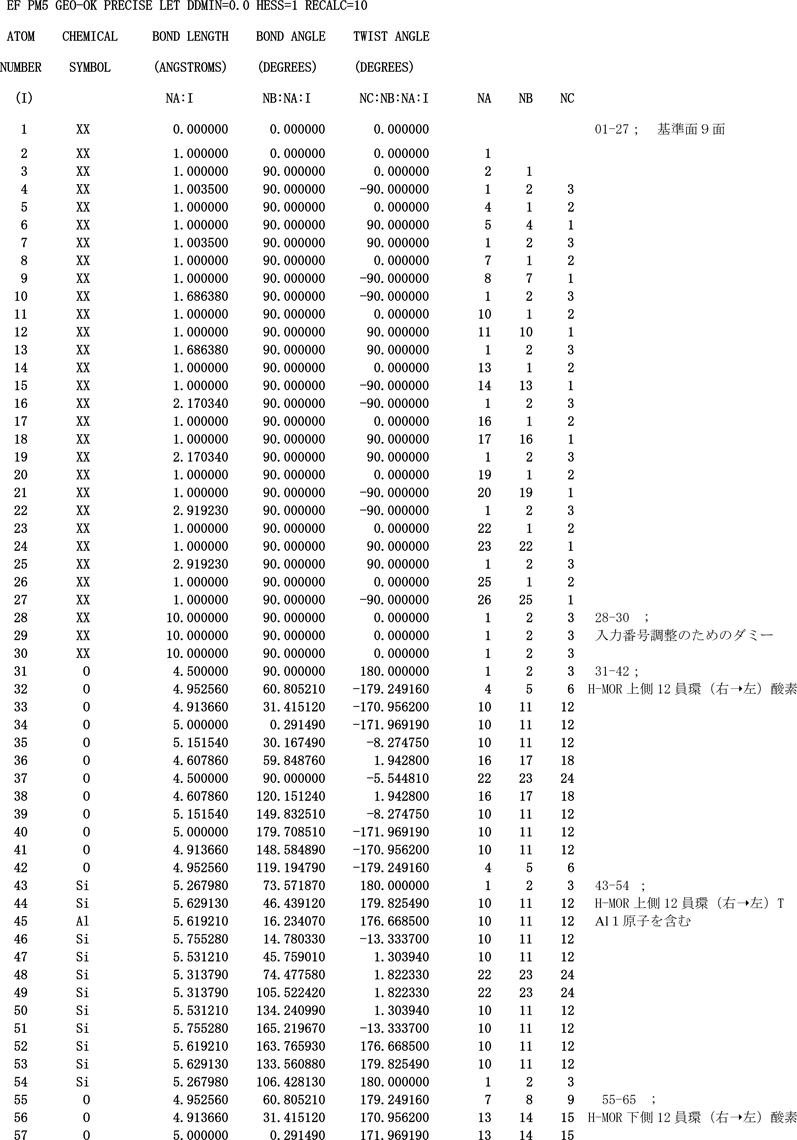
 |
6)操作1)と同様な基準面を用いて反応種(4-ペンテン酸)の構造最適化を別個に行い,最適化構造の座標値,ΔH0,HOMO,LUMOの各数値を求めた。
7)上記操作で得た反応種の最適化構造を,操作5)で得た構造データの下部,No.131以降の原子として追加入力した。この入力は,反応種を包接場中央付近に置くことになるが,No.131原子の可変要素すべて,No.132原子の結合長を除く可変要素二つ,およびNo.133の二面角可変要素の六つのみを可変とすると,配座を維持したままの反応種が,ΔH0が最小となるように細孔内を移動した。この時,操作5)で追加した細孔内の二つの水素を可変とすることもできる。この操作により,反応種の配座を固定したままではあるが,包接系に対するΔH0,HOMO, LUMOの各数値が求められた。
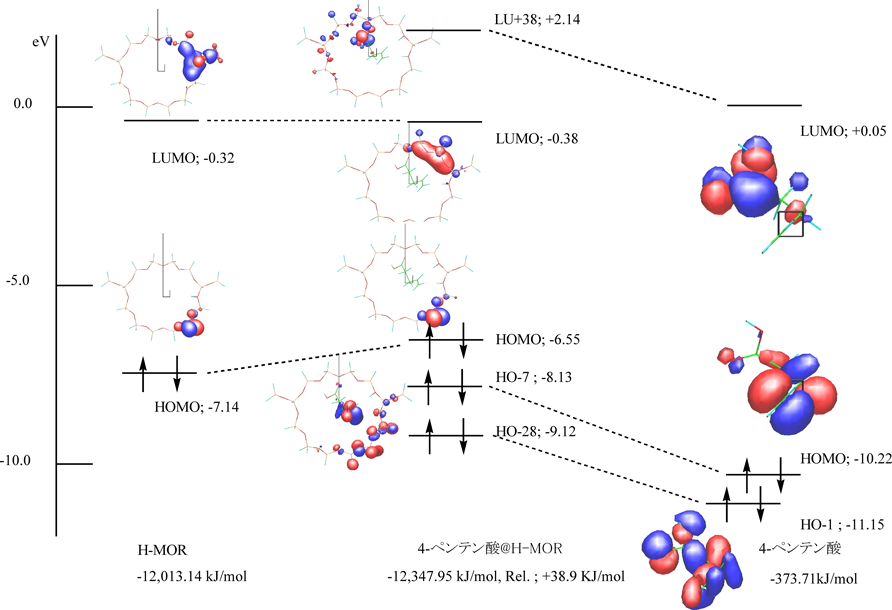
操作5), 6)および7)から得た結果を,図5の左から,包接場,反応種包接系,反応種の相関図として示した。これらの比較から次のような知見が得られる。H-モルデナイトのHOMOすなわち塩基点は,置換アルミニウム近傍の五員環部に分布する一方,LUMOは,アルミニウム置換に伴う負電荷を補償したB酸点付近に分布する。本系の分子内反応進行に欠かせないB酸点と塩基点との近接した位置関係は極めて興味深い。単独の包接場と反応種包接系とのB酸点の比較から,包接に伴うB酸点の転位が示唆された。包接場と反応種のΔH0値の単純和(−12386. 85 kJ/mol)と包接系に対する数値(−12347.95 kJ/mol)との比較から,この系の包接が38.9 kJ/mol吸熱的であることが示唆された。この結果は,アルケニルカルボン酸のH-モルデナイトに対する包接が,サイズ的には可能であるものの実際には起こりにくいという実験事実に一致する。反応種における占有軌道各々を各官能基に帰属した上でそれらの相対的準位を比較すると,オレフィン部位の電子供与能がカルボキシル基部位の供与能に優先することが示唆された。また,包接前後の各軌道のエネルギー準位を比較すると,包接場のB酸点に相当するLUMOに大きな変動がないのに対し,包接後の反応種アルケン部位のエネルギー準位が2.09 eV上昇した。このことは,アルケン部位から包接場のB酸点に対する電子供与能の増加が示唆され,包接場による反応促進効果が期待される。
ここまで示してきた包接場に対する半経験的分子およびそれらの包接系の軌道相関図軌道適用の検討は,緒に就いたばかりであるが,引き続き各種ゼオライトの基本骨格に対するデータベース化を進めると共に,3.2に示した各項目に対する高精度の検討を加え,本反応系を環境調和型有機合成法として確立していきたいと考える。併せて,この手法に対するご意見,ご批判を賜りたい。
ゼオライト包接下,アルキル介在鎖末端にカルボキシル基,エステル基,ニトリル基,ならびにアミド基を有するアルケンを反応種とする有用天然化合物ラクトンの環境調和型合成例を示した。併せて,本反応系の解析・設計を目的とした半経験的分子軌道法適用の可能性を示した。
最後に,本研究の遂行にあたり多大なご支援を戴いた日本化学工業株式会社に対して謝意を表したい。
引用文献References
1) J.-J. Filippi, X. Fernandez, L. Lizzani-Cuvelier and A.-D. Loiseau, Flavour Fragr. J., 21, 175-184 (2006).
2) M. F. Ansell and H. H. Palmer, Quarterly Rev., 18, 211-225 (1964).
3) Y. Zhou, L. Keith, W. Robert and J. Angelici, Applied Catalysis A General., 333, 238-244 (2007).
4) I.フレミング著,福井健一監修,竹内敬人,友田修司訳,フロンティア軌道法入門,講談社サイエンティフィク (1978).
5) 原伸宜,高橋浩,ゼオライト基礎と応用,講談社サイエンティフィク (1975).
6) 富永博夫,ゼオライトの科学と応用,講談社サイエンティフィク (1987).
7) 小野嘉夫,八嶋建明,ゼオライトの科学と工学,講談社サイエンティフィク (2000).
8) 特願2011-251995, 特願2011-251996.
9) W. Lutz, Microporous Mesoporous Mater., 90, 339-346 (2006).
10) 岡本康昭,表面科学,8, 596-602 (1991),岡本康昭,ゼオライト,10, 195-199 (1993).
11) 北川進,水野元博,前川雅彦,多核種の溶液および固体NMR, 三共出版,183-195, 313-319 (2008).
12) 山崎弘史,野村淳子,ゼオライト,28, 89-94 (2011), J. N. Kondo, D. Nishioka, H. Yamazaki, J. Kubota, K. Domen, and T. Tatsumi, J. Phys. Chem. C, 114, 20107-20113 (2010).
13) Y.-F. Li, J.-Q. Zhu, H. Liu, P. He, P. Wang, and H.-P. Tian, Bull. Korean Chem. Soc., 32, 1851-1858 (2011).
14) 平野恒夫,田辺和俊,分子軌道法MOPACガイドブック,海文堂出版 (1991).
15) シラノールから環状シロキサンに至る総合的な解説として,海野雅史,有機合成化学協会誌,69, 413-425 (2011).