近年,化石資源や化石燃料の利用による大気中の二酸化炭素の増加と地球温暖化問題の懸念から,循環型の低炭素社会の実現が望まれており,資源・エネルギーを有効利用する効率的な化学反応プロセスの構築,および二酸化炭素やバイオマスの再資源化など,グリーン・サステイナブルケミストリーを指向した革新的な化学技術の開発が求められている1)。硫酸や塩化アルミニウムに代表される酸触媒は,Fischerエステル化やFriedel-Craftsアルキル化反応など,各種の有機合成で古くから用いられているが,反応後に多量の塩が生成し反応の原子効率が悪いことや,反応後の触媒と生成物との分離操作が煩雑であること,触媒の回収・再使用が困難であること,などの問題があった。そのため,グリーン・サステイナブルケミストリーの観点からは,液相で機能する高効率かつ高選択的な固体酸触媒の開発が望まれている。
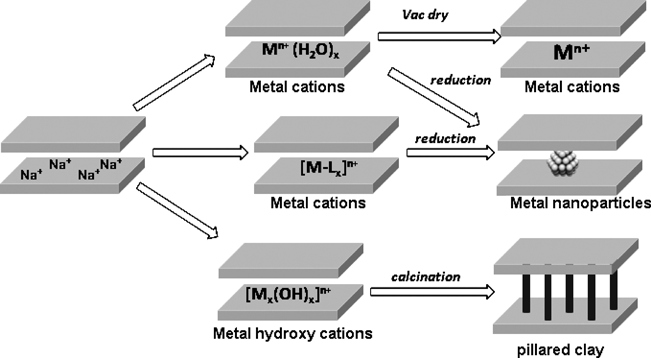
酸性層状粘土鉱物の一種であるモンモリロナイト(mont)は,2枚のシリカ四面体シートがアルミナ八面体シートを挟み込んだ2 : 1の基本層からなる。アルミナシート中のAl3+がFe2+やMg2+などの二価イオンに置換されることで,電荷を補償するために中間層にNa+やCa2+など対イオンを取込んだ積層構造をとる。この層間Na+が水や極性溶媒中では水和(溶媒和)することで,モンモリロナイトは非常に高い膨潤性を示し,有機反応における反応場として十分に機能する。また,層間Na+は,容易にH+や多価金属イオンと交換することができ,ピラーを立てるなどナノ空間を高度に利用する試みもなされている。さらには硫酸処理など新たなイオン交換能をもつ無機イオン交換体として利用されている(Fig. 1)2)。
筆者らのグループでは,層間Na+をH+や種々の多価金属イオン,有機アミンに交換したモンモリロナイトを調製し,液相での各種有機合成反応の優れた固体酸触媒となることを報告してきた3)。層間内での金属イオン種の形態は,金属イオンの加水分解定数に関係しており,金属カチオンの種類によって,鎖状酸化物集合体やサブナノ金属クラスター,単核金属種を作り分けることができ,それぞれ特徴的な反応が進行する。本稿では,グリーン・サステイナブルケミストリーを指向する有機合成反応,なかでもモンモリロナイトを固体ブレンステッド酸触媒として用いる炭素–炭素結合形成反応,炭素–窒素結合形成反応および炭素–酸素結合形成反応を例として筆者らのグループの最近の例を述べる。
2. プロトン交換モンモリロナイト(H-mont)触媒
2.1. 炭素–炭素結合形成反応
炭素–炭素結合形成反応は,ファインケミカルズ,バルクケミカルズを問わず,新たな分子骨格を構築するために極めて重要な反応である。例えば,フリーデルクラフツ反応のように,ハロゲン化アルキルを試剤とするアルキル化反応や,硫酸などの液体酸をブレンステッド酸触媒として用いた反応では,反応後の中和処理によって多量の塩が副生することや,生成物の選択性制御が困難であるなどの問題点があった。近年では,金属錯体を用いた中性条件での選択的炭素-炭素結合形成反応が広く研究されているが,反応後の触媒と生成物との分離が困難といった課題がある。
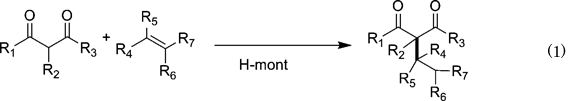
Na-montを塩酸水溶液で処理すると,mont層間にH+を導入したプロトン交換モンモリロナイト(H-mont)を調製することができる。このH-montは,優れた固体ブレンステッド酸触媒となり,不活性な単純オレフィンへの1,3-ジカルボニル化合物の付加反応が効率よく進行する(Eq. 1)4)。
従来の酸塩基触媒を用いる1,3-ジカルボニル化合物のオレフィンへの付加反応では,使用できるアクセプターはα,β-不飽和ケトン等の活性オレフィンに限定されていた5)。種々の酸触媒を用いてアセチルアセトンのノルボルネンへの付加反応を行ったところ,H-montを用いると収率90%で相当する付加体が得られた(Table 1)。固体酸として知られるHUSYの触媒活性は低く,酸強度の大きいHmordenite,細孔径の大きいAl-MCMや均一系の硫酸等を用いても反応はほとんど進行しない。Hmontを用いる反応系では,ジケトンだけでなく,ケトエステルの反応にも適応可能である。さらに,ノルボルネン以外にも1-ヘキセンやシクロヘキセン,スチレンといった単純オレフィンへの付加反応も進行する。H-mont層間に濃縮されたブレンステッド酸点がオレフィンとジカルボニル化合物の両方を活性化し,これまで困難であった単純オレフィンへの付加反応が進行すると考えられる(Table2)。
Table 1. Addition of 1 to 2 using various acid catalystsa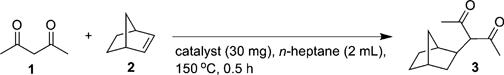 |
|---|
| Entry | Catalyst | Ammount of acid site (mmol/g cat)b | Pore size (Å) | Conv. 1 (%)c | Yield 3 (%)c |
|---|
| 1 | H-mont | 0.86 | — | 87 | 83 |
| 2 | H-USY | 0.53 | 7.4×7.4 | 37 | 37 |
| 3 | mont K-10 | 0.25 | — | 9 | 8 |
| 4 | Al-MCM-41 | 0.15 | 27×27 | 7 | 5 |
| 5 | H-mordentie | 1.07 | 6.5×7.0 | 11 | trace |
| 6 | H-ZSM-5 | 0.87 | 5.1×5.5 | n.r. | — |
| 7 | SO42−-ZrO2 | n.m. | — | n.r. | — |
| 8 | Na+-mont | n.m. | — | n.r | — |
| 9d | H2SO4 | — | — | 29 | 4 |
| 10 | p-TsOH | — | — | n.r | — |
| 11 | none | — | — | n.r | — |
| a Reation conditions: 1 (1.3 mmol), 2(1.0 mmol). b Estimated by NH3-TPD. c Determined by GC and 1H-NMR. d H2SO4 (0.1 mmol). n.r.=no reaction, n.m.=not measured. |
Table 2. Addition of 1,3-dicarbonyls to alkenes catalyzed by H-monta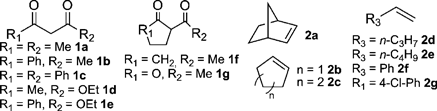 |
|---|
| Entry | 1 | 2 | Time (h) | Temp. (°C) | Yield (%)b |
|---|
| 1 | 1a | 2a | 1 | 150 | 90 |
| 2 | 1b | 2a | 1 | 150 | 88 |
| 3 | 1c | 2a | 1 | 150 | 93 |
| 4 | 1d | 2a | 1 | 150 | (71) |
| 5 | 1e | 2a | 1 | 150 | 74 |
| 6 | 1f | 2a | 1 | 150 | 83 |
| 7 | 1g | 2a | 1 | 150 | 87 |
| 8c | 1c | 2b | 24 | 180 | (66) |
| 9c | 1b | 2c | 24 | 180 | 78 |
| 10c | 1c | 2c | 24 | 180 | (80) |
| 11d | 1c | 2d | 24 | 150 | 53 |
| 12d | 1c | 2e | 24 | 150 | 60 |
| 13e | 1e | 2f | 3 | 150 | 72f |
| 14g | 1f | 2g | 3 | 150 | 72f |
| a Reaction conditions: 1 (1 mmol),2 (1.3 mmol), Hmont (0.115 g), n-heptane (2 mL).b Isolated yield based on 1. Values in parentheses are GC yield.c 2 (5 mmol).d 2 (15 mmol).e 1e (3 mmol), 2f (1 mmol).f Isolated yield based on 2.g 1f (1.3 mmol), 2g (1 mmol). |
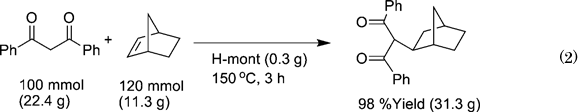
反応後の触媒は,遠心分離やろ過により容易に回収が可能であり,活性および選択性の低下なく7回の再使用が可能であった。また,100 mmolスケールのノルボルネンとジベンゾイルメタンとの反応は,H-mont(0.3 g)を用いると3時間でほぼ完了する(Eq. 2)。
反応終了後,生成物は溶媒であるn-ヘプタンから再結晶で容易に単離が可能である。この反応においてH-montの酸点あたりのTONは327, TOFは109h−1となり,これまでに報告された触媒系にくらべ非常に高い値である。本反応は,均一系試剤である硫酸やp-トルエンスルホン酸を用いても全く進行しない。このH-montは,基質と求核剤の両方を活性化(dual activation)できるため,ユニークな触媒作用が発現したと考えられる。
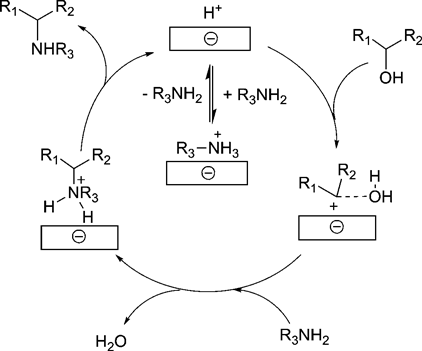
H-montのブレンステッド酸点は,アルコールをアルキル化試剤とする1,3-ジカルボニル化合物のアルキル化においても高い触媒活性を示す6)。求核置換反応にはハライドやアセテート,およびカーボネートを用いた方法が一般的であるが,廃棄物として塩が生成する問題があった。アルコールを用いた方法は水のみを副生することから理想的であるが,反応性が低いため一般的に困難であり,当量もしくはそれ以上の試剤が必要であった。最近では,均一系酸触媒として,NaAuCl4, InCl3, ZrCl4,(La, Yb, Sc, およびHfの)金属トリフラート,B(C6F5)3, BF3, およびp-トルエンスルホン酸を用いたアミド7),1,3-ジカルボニル化合物8),およびアリルシラン8d,9)による,アルコールの求核置換反応が報告されている。しかし,これらの触媒は触媒活性および選択性が低いこと,再使用が困難であることや環境に優しくないハロゲン溶媒の使用が必要であるなどの問題があった。これに対して,H-montを用いた反応系では,種々のベンジル型およびアリル型アルコールの水酸基が効率的に脱離基として機能し,1,3-ジカルボニル化合物のアルキル化が進行する(Table 3)。上述のオレフィンのアルキル化と同様に,層間のブレンステッド酸点でのdual activationにより効率的に反応が進行している(Scheme 1)。
Table 3. Benzylation of allylation of 1,3-dicarbonyls with alcoholsa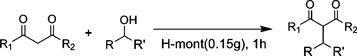 |
|---|
| Entry | 1 | Alcohol R, R' | Temp. (°C) | Yield (%)b |
|---|
| 1 | 1a | Ph, Me | 100 | 90 |
| 2 | 1a | 4-Cl-Ph, Me | 150 | 80 |
| 3c | 1a | 2naphthyl, Me | 150 | 72 |
| 4 | 1a | Ph, Ph | 100 | 91 |
| 5 | 1a | 2-cychlohexenol | 90 | 80d |
| 6e | 1d | Ph, Me | 100 | 86 |
| 7c | 1f | Ph, Me | 150 | 86 |
| 8 | 1d | Ph, Me | 100 | 88 |
| 9 | 1e | Ph, Me | 100 | 51 |
| 10 | 1d | 2-cyclohexenol | 90 | 48 |
| a Reaction conditions: 1 (1.5 mmol), 2 (1 mmol), H-mont (0.15 g) , n-heptane (2 mL). b Isolated yield based on alcohol. c Time 0.5 h. d GC yield. e 1b (1 mmol). |
2.2. 炭素–窒素結合形成反応
上述のアルキル化反応では,H-montのブレンステッド酸点が特異な活性,選択性に寄与し,新たな炭素–炭素結合が生成した。このようなアルコールの脱水によるカルボカチオン生成と求核剤との反応を利用すると,種々の炭素-窒素結合形成反応への応用が可能となる。H-montは,アリル型アルコール,ベンジル型アルコールを用いたアニリン(Table 4)やスルホンアミドのように塩基性の低い窒素求核剤のアルキル化にも高活性を示す10)。シクロヘキセノールを用いたアニリンのアルキル化では,ゼオライト系触媒に比べてH-montが優れた機能を発揮する。H-betaやH-USYも良好な収率を与えるが,H-ZSM-5や濃硫酸,p-トルエンスルホン酸は低活性であった。また,p-トルエンスルホンアミドを求核剤とする場合にも効率良く反応が進行し,各種のベンジル型アルコールにより炭素–窒素結合が生成する。これらの窒素求核剤を用いた炭素–窒素結合形成反応においても,先述の炭素–炭素結合形成と同様に,層間のブレンステッド酸点による基質のdual activationによって反応が進行すると考えられる(Scheme 2)。
Table 4. Benzylation of anilines catalyzed by H-monta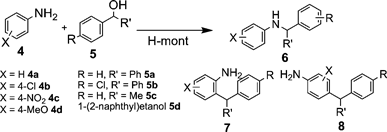 |
|---|
| Entry | Amine | Alcohol | Time (h)/temp (°C) | Yield (%)b | 6 : 7 : 8c |
|---|
| 1 | 4a | 5a | 1/150 | 96 | 51 : 0 : 49 |
| 2 | 4b | 5a | 3/150 | 83 | 100 : 0 : 0 |
| 3 | 4c | 5a | 1/100 | 97 | 100 : 0 : 0 |
| 4b | 4c | 5a | 1/100 | 98 | 100 : 0 : 0 |
| 5 | 4d | 5a | 30/150 | 39 | 100 : 0 : 0 |
| 6 | 4c | 5b | 1/100 | 93 | 100 : 0 : 0 |
| 7 | 4a | 5c | 15/125 | 93 | 54 : 28 : 18 |
| 8 | 4c | 5c | 1/100 | 77 | 100 : 0 : 0 |
| 9 | 4a | 5d | 30/100 | 99 | 82 : 9 : 18 |
| 10 | 4c | 5d | 1/100 | 93 | 100 : 0 : 0 |
| a Reaction conditions: 4 (2 mmol), 5 (1.0 mmol), H-mont (0.1 g), 1,4-dioxane (2 mL). b Isolated yield. c Determined by GC. d Third reuse experiment. |
3. Tiイオン交換モンモリロナイトによる炭素–酸素結合形成反応(グリセロールの選択的アセタール化)
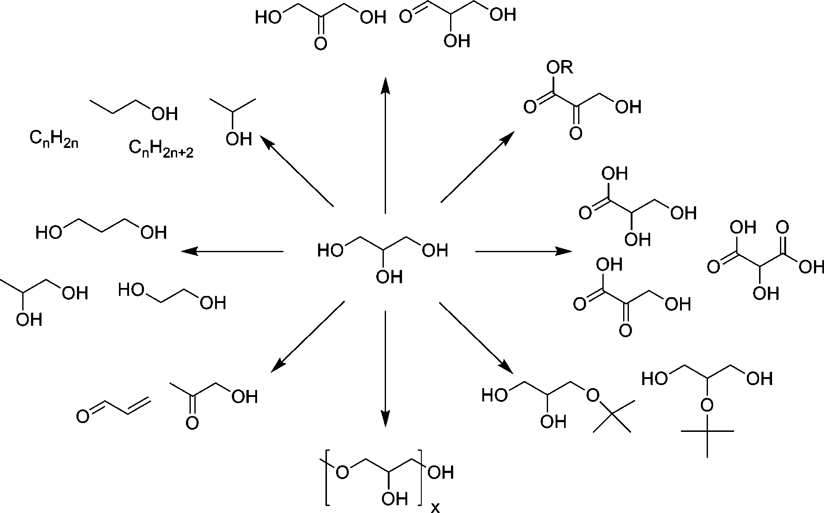
バイオマス資源の一つであるグリセロールは,最も単純な三炭糖(トリオース)であり,主に石鹸や油脂工業における副生物として得られる。最近では世界的なバイオディーゼルの生産増加により,大幅にその生産量が増加している。それに伴い,グリセロールからの種々の有用化合物への高度変換は盛んに研究されはじめているが(Scheme 3),未だ多くのグリセロールは焼却処理しているとされ,有用物質への高選択的変換法の確立が望まれている11)。
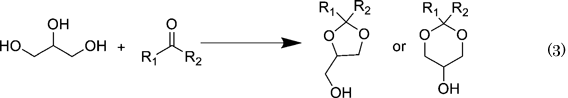
グリセロールの誘導体化法として,酸触媒を用いグリセロールのヒドロキシル基をケトンやアルデヒドと反応させアセタール化する手法がある(Eq. 3)12)。アセタール化することで,3つの水酸基のうち2つのヒドロキシル基を保護し,さらに残り1つのヒドロキシル基を選択的に反応させることが可能になる。また,有機溶媒への溶解性が向上しグリセロールに比べて非常に扱いやすくなるという利点がある。これらのアセタールはバイオディーゼルやガソリンの添加剤としても有用である。
Tiイオン交換モンモリロナイト(Ti-mont)は,Na-montを四塩化チタン水溶液で処理して調製した3f)。Ti-montの層間には鎖状水酸化物種が生成し,架橋酸素上のプロトンが強いブレンステッド酸性を発現し,アルキル化反応やエステル化,アセタール化,脱アセタール化などに優れた固体触媒として機能する3f,13)。Ti-montを触媒として,アセトンを自己溶媒に用いグリセロールと反応させたところ,収率>99%で5員環アセタール化物(1)が得られた(Table 5)14)。強酸性ゼオライトとして知られる,H-ZSM-5やH-Betaでは,反応はあまり進行しない。同様に超強酸性イオン交換樹脂であるNafion-H NR50では収率7%,市販の硫酸処理されたモンモリロナイトであるK-10では5%とほとんど反応が進行しなかった。また,H-Yゼオライトを触媒とした場合では,まったくアセタール化反応を触媒せず,Ti-montはグリセロールとアセトンからの環状アセタール合成反応に対し,穏和な条件で従来の固体酸触媒に比べ非常に高い触媒活性を示した。
Table 5. Acetalization of glycerol with acetone using various acid catalystsa |
|---|
| Entry | Catalyst | Time (min) | Yield of 1 (%)b | Yield of 2 (%)b |
|---|
| 1 | Ti-mont | 6 | 88 | 5 |
| 2 | Ti-mont | 15 | >99 | <1 |
| 3 | Ti-mont (reuse 1) | 6 | >99 | <1 |
| 4 | Ti-mont (reuse 2) | 6 | >99 | <1 |
| 5 | Ti-mont (reuse 3) | 6 | 99 | 1 |
| 6 | H-ZSM-5 | 6 | 17 | 5 |
| 7 | H-Beta (Si/Al=12.5) | 6 | 16 | 4 |
| 8 | Nafion NR50 | 6 | 7 | 3 |
| 9 | K-10 | 6 | 5 | 2 |
| 10 | HY | 6 | 0 | 0 |
| a Reaction conditions: catalyst (0.05 g), glycerol (5 mmol), acetone (4 mL), room temp. b Determined by GC-MS using an internal standard method. |
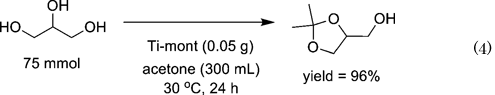
Ti-mont触媒の再使用性の検討を行ったところ,触媒活性・選択性の低下なく少なくとも3回の再使用が可能であった。グリセリン75 mmolを用いたラージスケールでの反応においても,30°C,空気中で24時間攪拌したところ単離収率96%で5員環アセタール化物が得られた(Eq. 4)。
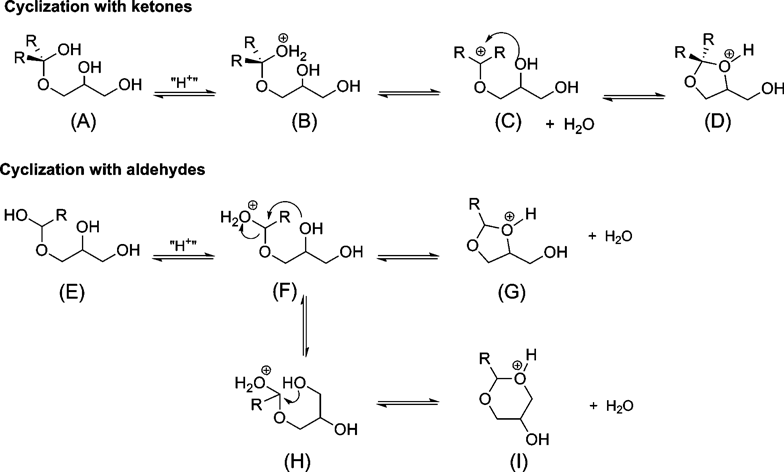
グリセロールと種々のカルボニル化合物との反応を行ったところ,Table 6に示すようにケトンおよびアルデヒドで相当するアセタールが高収率で得られた。ケトンとの反応では高選択的に5員環アセタールが得られる。これは,ケトンとの反応においては,へミアセタールからの脱水により,3級のカルベニウムイオン(C)が生成し,その後,素早く2級のヒドロキシル基が求核攻撃することで,速度論的に優先する5員環アセタールが選択的に生成するためと考えられる(Scheme 4)。一方,アルデヒドを用いた場合は,(C)に比べて(F)の寿命が長いため,速度論的に優先な5員環アセタールだけでなく熱力学的に安定な6員環アセタールも生成する。
Table 6. Synthesis of cyclic acetals of glycerola |
|---|
| Entry | Substrate | Time (min) | Yield (3+4) (%)b | Ratio (3 : 4)b |
|---|
| 1 |  | 15 | 99 | 98 : 2 |
| 2 | 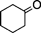 | 1 | 99 | 99 : 1 |
| 3 | 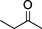 | 0.5 | 99 | >99 : 1< |
| 4 | 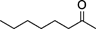 | 8 | 95 | 99 : 1 |
| 5 | 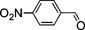 | 15 | 97 | 66 : 34 |
| 6 | 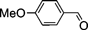 | 8 | 99 | 42 : 58 |
| 7 |  | 1 | 94 | 60 : 40 |
| a Reaction conditions: glycerol (1 mmol), carbonyl compounds (2 mmol), Ti-mont (0.1 g), MS4A (0.5 g), 1,4-dioxane (5 mL), room temp. b Determined by GC based on glycerol. |
H-mont やTi-mont は高選択的なC–C, およびC–N, C–O結合形成反応に優れた固体酸触媒として機能することを見出した。これらの触媒は,環境に優しい天然の層状粘土であり,適応した反応のみならず,触媒自体も環境に優しい化合物と言える。今後,モンモリロナイトでは,オングストロームスケールの層間内酸点の強度や量の制御,層を構成するシリケート層との協奏機能など,新たな触媒設計と新規反応の開拓を行いたい。