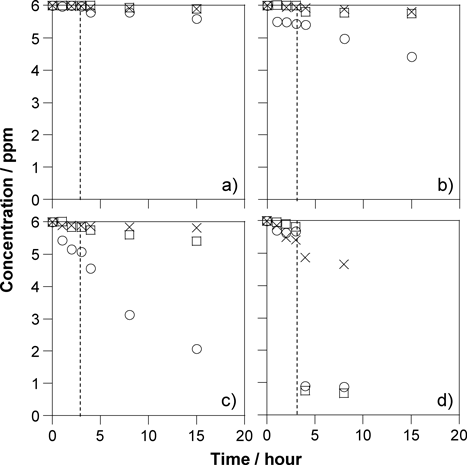
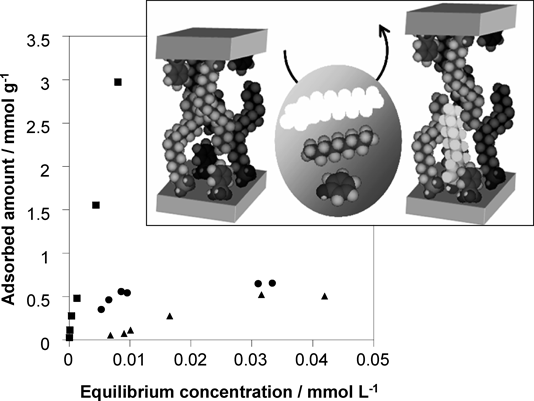
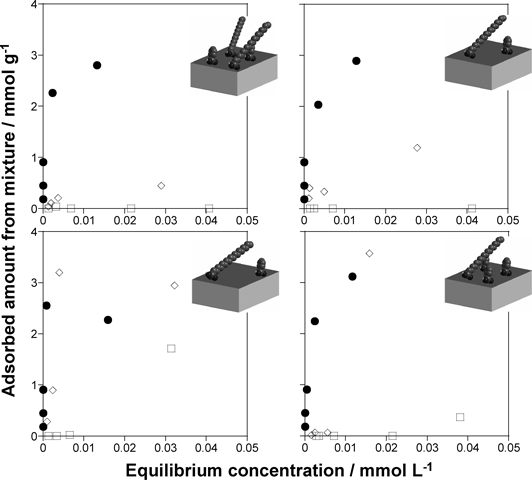
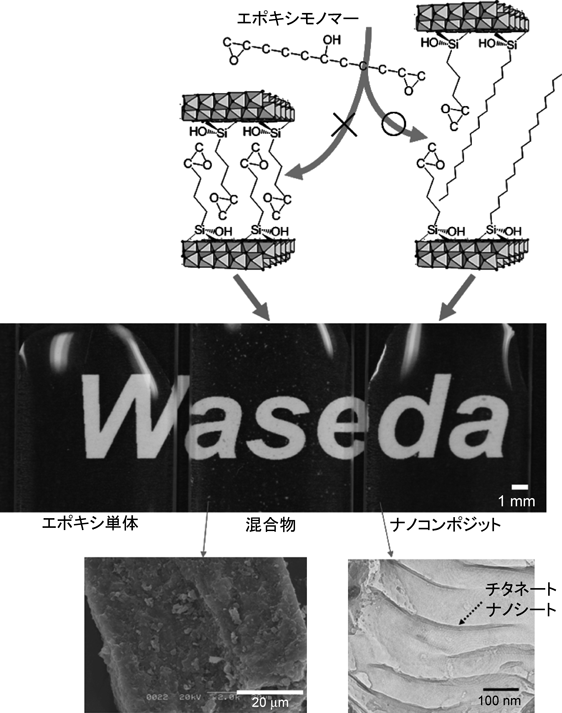
層状チタン酸塩の機能化Functionalization of Layered Titanates
1 広島大学大学院工学研究科応用化学専攻Department of Applied Chemistry, Graduate School of Engineering, Hiroshima University ◇ 〒739-8527 広島県東広島市鏡山1-4-1
2 早稲田大学教育・総合科学学術院地球科学専修Department of Earth Sciences, Waseda University ◇ 〒169-8050 東京都新宿区西早稲田1-6-1
受理日:2012年6月29日Accepted: June 29, 2012
発行日:2012年9月7日Published: September 7, 2012