合成法は三つに大別できる。第一の方法は,直鎖アルキル基と親水基からなる界面活性剤分子のミセルを利用する方法である。親水基にはスルホ基,スルフェート基,アミノ基,カルボキシル基,リン酸基が考えられるが,これらはメソ細孔性金属酸化物合成の研究初期に広く試されたため,チタニアに関しても全て利用が試みられたと思われる6)。アルキルリン酸では型剤除去後に良好な周期性メソ細孔構造が得られたが,メソ細孔性チタニア中にリンが残留することが明らかにされている。結局この型の界面活性剤では一級アミンを利用すると合成が容易になるようで,チタン源はアルコキシドでよい7)。型剤分子がMCM-41やSBA-1の合成で用いられる四級アンモニウムと異なる理由は,シリカとチタニアでは水熱合成時の前駆体の電荷密度が異なるためであろう。この方法で得られる固体のBET比表面積は700–1200 m2 g−1にも達する8)。最も解明されているチタニア単結晶表面はrutile (110)であるが,この単結晶表面の原子層だけからなる仮想的な固体の表面積は1440 m2 g−1となる。この数字と比較すると1200 m2 g−1という表面積が非常に大きいことが実感できる。一方四級アンモニウムを用いても合成は可能で,蒸発誘起自己集合体形成(evaporation-induced self-assembly, EISA)法の条件を最適化することにより比表面積280–370 m2 g−1のメソ細孔性チタニアが得られる9)。またチタン前駆体として過硫酸塩(TiOSO4・xH2SO4・xH2O)を用い,比表面積262 m2 g−1のメソ細孔性アナターゼも得られている10)。他にもn-アルキルスルフォネート12)を型剤に良好なメソ細孔構造を有するチタニアが得られる。ただしTi(III)塩を型剤にする11),注意深く酸化還元雰囲気の調整する12)などの工夫が必要である。
第二の方法はブロック共重合体を用いる合成法で,ここで分類した三つの方法の中では最も広く行われている。型剤はPluronic共重合体(PEOn-PPOm-PEOn)が一般的で,チタン前駆体には塩化物やアルコキシドが利用される13)。この方法では溶液で試薬を混合し,乾固させて周期性メソ構造を形成させた後に,主に焼成で型剤を取除くという手順になり,基板上にメソ細孔性チタニア薄層を形成させる手段として都合が良い。P12314–18),F12715,16,19)などのPluronic 共重合体以外にも,Brij 5815,19),Brij 5615),クレイトン液-ポリオキシエチレン(PHB-PEO, Mw=7480)20)などのアルキルポリオキシエチレン共重合体(R-PEOn),ポリスチレン-ブロック-ポリオキシエチレン共重合体(PSmb-PEOn)21),Triton X-10022)などのポリオキシエチレン-アルキルフェニルエーテル(R-Ph-PEOn)を用いた合成例がある。
第三の方法は無機鋳型を用いる合成法である。メソ細孔性シリカ23,24)やメソ細孔性炭素24)の細孔中へチタンアルコキシドの溶液を充填し,熱分解,酸化した後,型剤を焼成または溶解する。ただしシリカを型剤にした場合は,ケイ素の完全除去は困難で,良好な周期構造を与えるSBA-15型剤の場合でSi/Ti>3 atom%のケイ素が残留する。SBA-16, FDU-12では良い周期構造は得られない24)。
3.1 細孔構造
n-アルキルアミンやn-アルキルスルフォネートが型剤の場合,細孔径は2.5 nm-4 nm程度で7,8),ブロック共重合体を型剤とした時はそれより大きくなる。(Pluronic P123で6.5 nm程度13),PHB-PEOで10 nm程度20),PS-b-PEOで数十nm。)21)n-デシルアミン(C120-NH)からn-オクタデシルアミン(C18-NH2)までの炭素数10, 12, 14, 16, 18の型剤でメソ細孔性チタニアを合成したところ,細孔径はアルキル鎖長を反映して2.6 nmから3.7 nmまで変化する8)。これはn-デシルトリメチルアンモニウム(C10-N+Me3)からn-オクタデシルトリメチルアンモニウム(C18-N+Me3)まで型剤を変えて合成されたMCM-41の細孔径の変化(1.9-3.2 nm)と同程度である25)。ブロック共重合体を利用する合成法では,PS40000-b-PEO53000からPS100000-b-PEO150000の間で分子量を変え,細孔径を40から100 nmまで変化させた研究例がある21)。
シリカに比べると周期構造の規則性が高いチタニアは得にくく,また周期構造の多様性もシリカより劣る。n-アルキルアミンやn-アルキルスルフォネートを型剤とした場合,得られる粉末X線回折の低角領域での回折線は通常一つだけである。これはメソ細孔の位置相関の規則性を示しており,いわゆるwormhole-like構造に帰属される。さらにこのピークさえ出現しない場合も多い。しかしながら細孔径分布は依然として十分狭い場合も多い8,11,25)。型剤除去前では二次元六方晶(p6mm)に帰属しうる高次回折線が出現することがあり7,26,27),この場合,除去後には高次回折線が幅広になったと考えて矛盾しないことから7,9,10),このカテゴリーの型剤を用いた場合は,直管状の均一なメソ細孔が二次元六方対称に配列した状態からやや乱れた構造になっていると考えられる。
ブロック共重合体や無機鋳型を用いて調製された場合,細孔径分布は狭くともX線回折で明確な高次構造が出現しないことが圧倒的に多い。基盤上に展開される薄層であることが多く,電子顕微鏡で構造解析されることが多いが,そのレベルでは二次元六方晶15,19,20),三次元六方晶(P63/mmc)17),立方晶(Im3m)15,20),が確認されている。
3.2 結晶構造
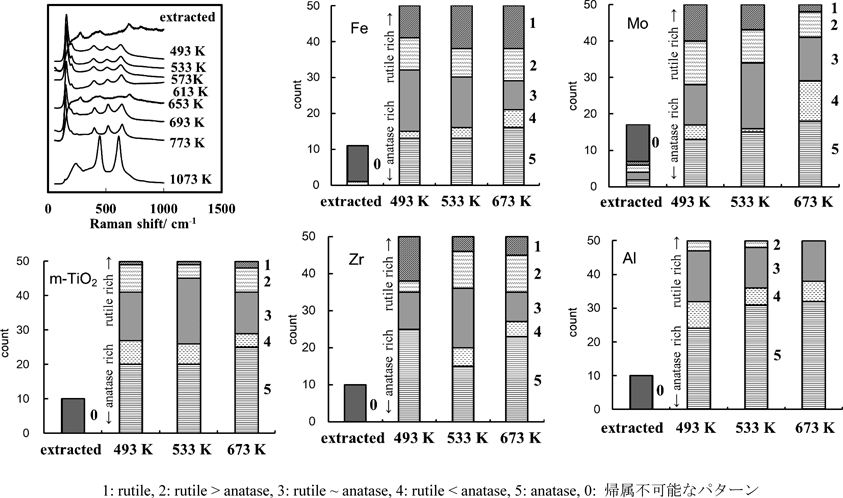
n-アルキルアミンを型剤とする合成法では,生成するチタニアは結晶性に乏しい8,27)。このことは大きなBET比表面積と符合する。これを大気中で加熱することにより結晶化するが,相転移温度,相転移後の結晶相,相転移後のメソ構造は条件,添加物に依存する。Tiに対してモル比で1%のCe4+, Nb5+,Al3+, Fe3+, Zr4+, Mo5+を加え,C12-NH2を型剤に直接合成したメソ細孔性チタニア29,30)では,無添加のメソ細孔性チタニアと同様,380–400°Cの大気下焼成でアナターゼに変化する。またこの変化に伴いミクロ孔の大幅な減少,細孔の拡大(2.0 nmから添加物に依存し3.5–4.0 nmへ。細孔径分布の幅は常に狭い。),wormhole-like構造に帰属されるX線回折線の消失が見られた。換言すると非晶質-アナターゼの結晶相の転移に伴い,メソ細孔構造も転移を起こす。これより低温度での焼成後のメソ細孔性チタニアの構造は回折線には変化がなく,振動スペクトルのパターンのみ変化する。焼成温度を変え,一温度あたり50点ずつラマン散乱を記録すると,すべての試料で200°C以下の低温焼成ではバンドがほとんど現れないが,それ以上の温度ではアナターゼとルチルのパターンが観測される。これらのスペクトルは340–380°Cで消失,380–400°Cでの結晶化後はアナターゼのスペクトルのみが得られることが明らかになった(図1)。転移温度以下の中間温度でルチルとアナターゼのスペクトルが同じ試料で観測されることは,アナターゼやルチルの微結晶が同時に生成することか,局所的に原子配列がそれらの構造になることを示すが,アナターゼへの結晶化の直前の温度で,ラマンバンドがすべて消失するため,微結晶の生成ではなく局所的な原子配列の変化であると考えられる。200–340°Cの温度で見いだされるアナターゼ,ルチルのラマンバンドの出現頻度およびその温度変化は添加物に大きく依存する。Zr4+, Mo5+の場合,温度上昇とともにルチルの相対頻度が減少,Fe3+, Nb5+の場合,温度上昇とともにルチルの相対頻度が増大,Ce4+,Al3+の場合,ルチルのピークの出現は比較的まれである。(図1)非晶質メソ細孔性チタニアの原子配列は,僅かな(Ti/M=100)添加物によって大きく影響を受けることが明らかになった。
他にミクロ細孔性チタニアの(試料中複数の場所ではなく)一カ所でラマンスペクトルを測定した研究例で,ルチルのラマンバンドが出現する温度より高い温度で焼成した後,アナターゼのバンドが得られることが報告されている31)。
チタンの過硫酸塩とCTABを用いた合成10),四塩化チタンとn-ドデシルスフォネートを用いた合成11)では,それぞれアナターゼ,ルチルの回折線を示すメソ細孔性チタニアが得られる。詳細は不明であるが,これらの結晶相の形成に硫酸根が何らかの効果を及ぼしている可能性がある。
ブロック共重合体を用いた合成の多くは,400°C程度で焼成して型剤を取除くが,生成する相は主にアナターゼである。しかしPluronic P123を型剤に形成したメソ構造体を窒素中350°Cで加熱すると非晶質のままp6mmのメソ細孔性固体に変化する。これを酸化するとX線の高次回折線は消失し,メソ細孔性アナターゼ変化する28)。同じ界面活性剤を用いても合成条件次第ではアナターゼ,ルチル,ブルカイトの混相がさまざまな割合で生じる14,16,32)。また TiO2(B)33)に帰属される結晶相を有するメソ細孔性チタニアも報告されている18)。
3.3 局所構造
結晶化しない低温で焼成した非晶質メソ細孔性チタニアがアナターゼやルチルのラマンスペクトルを与えることは,既述のとおりである。これは局所構造に他ならないが,吸着,触媒,電極など,チタニアに期待される多くの応用が表面構造に敏感であることを考えると,詳細な局所構造の解析は重要である。
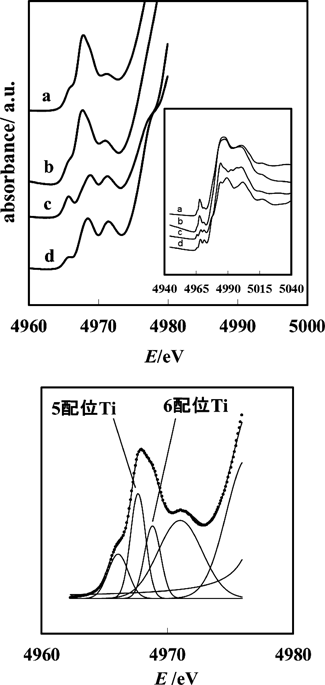
26種類の結晶性酸化物を対象に,チタンK吸収端のXANESのpre-edgeピークの位置や強度が,Ti原子に配位する酸素原子の数により異なることが解明されている34)。アナターゼもルチルも6配位チタンのみ存在し,表面に限って5配位,4配位が出現する35)。したがってこれらの結晶の透過スペクトルを測定しても,通常6配位のpre-edgeのみ観測される。一方n-ドデシルアミンを型剤に合成されたメソ細孔性チタニアのTi K吸収端pre-edgeでは,5配位に帰属されるピークが主要なピークであり,6配位に帰属されるピークが分離されずに出現する(図2)36)。ピーク分離によって5配位対6配位の濃度比を見積もったところ,2 : 3となった。このメソ細孔性チタニアは1200 m2 g−1を超える比表面積から表面の割合が非常に高いと予想されるが,この予想は異常に高い配位不飽和性と符合する。Ti K吸収端pre-edgeは573 K焼成ではほとんど変化が見られず,アナターゼへの結晶化が起きる673 Kの焼成後はアナターゼのスペクトルに変化する36)。
配位不飽和性が高い表面は反応性に富む反面,不安定になるが,チタンアルコキシドのCVDとその後の酸化によって比表面積がほとんど低下することなく,表面が安定化する8)。この方法の場合,N, P, Sなどの異種元素の添加がないことは純物質の調製という観点からは長所である。
ドデシル硫酸を型剤にメソ細孔性アナターゼを合成する途中段階,焼成前ゲルのTi K吸収端EXAFSスペクトルからは,Ti–S結合の存在が示されている12)。
還元処理後のチタニアからESR信号が得られることは古くから知られる37)。メソ細孔性チタニアも格子欠陥が多く,ESRによっていろいろな構造情報が得られるのだが,液体ヘリウム温度程度の低温が必要であり,メソ細孔性チタニアの構造解析に適用された例は少ない。水の吸着によって導電帯の電子が検出できなくなることなどが明らかにされている38)。
光触媒作用の研究は多い10,14,18,26,27,32,39–41)。大部分が光照射による有機分子の酸化反応で,非細孔性チタニアと同様,結晶相がアナターゼあるいは主としてアナターゼのメソ細孔性触媒が研究されているが,Xeランプ照射下におけるメソ細孔性TiO2(B)によるアセトン酸化がP-25(anatase : rutile〜4 : 1の組成を持つ,比表面積が約50 m2 g−1のチタニア・エアロゾル)よりも2倍以上活性であることも報告されている18)。メソ細孔性アナターゼの場合は,P-25と比較して活性が高い場合14,26)もあれば,低い場合27,32)もある。メソ細孔性チタニアの研究では,均一な細孔直径を変化させて合成できるため,拡散と活性の関係について探求でき,反応機構解明の点から注目される39)。
n-アルキルアミン型剤から合成されるメソ細孔性チタニアは表面積が大きいが,400°C以上で焼成しない限り非晶質なため,光触媒ではなく,配位不飽和性が高い触媒担体としての関心が高い29,41,42)。触媒成分が単核で存在するような遷移金属酸化物触媒については,酸化数,配位数が既存のチタニア担体を利用した場合とは大きく異なることがある。Ⅴ担持触媒ではその調製が直接合成法か含浸担持法によって詳細が異なるが,Ⅴの酸化数は従来のチタニア担体触媒よりも低くなる41)。直接合成触媒V-meso TiO2では大気中のV K吸収端XANESスペクトルのpre-edgeピーク位置がV4+の位置と一致する。一方,担持触媒V/meso TiO2やP-25に担持した触媒V/P-25ではV5+の位置にピークが出るが,前者のpre-edgeピークの半値全幅は大きい。またこれらのメソ細孔性チタニア担持V触媒は,室温大気中でも明快なV4+のESR信号が観察される。アナターゼ担体,ルチル担体では室温大気下,Ⅴ触媒のESRは出現しないことから,メソ細孔性チタニア担体ではV4+が異常に安定化されていることが明らかである。バナジウム触媒の接触酸化では,Ⅳ-Ⅴ価間のredox機構が主要な機構とされているので,V4+の安定化は触媒作用を大きく変化させる。200°C真空脱気前後で,V-meso TiO2のスペクトルではB=(g║−ge)/(g⊥−ge)がほとんど変わらないのに対して,V/meso TiO2では大きく減少する。(表1)このパラメーターはⅤのtetragonal distortionの指標となるとされており,後者の触媒は水の吸着でVの配位環境が大きく変化していることを示唆する。V4+の安定性と存在割合は,V-meso TiO2とV/meso TiO2で異なることがそれぞれのESRスペクトルの比較,それぞれのESRとXANESとの比較からわかる。触媒燃焼活性は,VP25との比較で前者の触媒が7.6倍,後者の触媒が23倍になる41)。
表1 1.4 wt% V-meso TiO2および1.3 wt% V/meso TiO2のⅤ(Ⅳ)の電子スピン共鳴構造定数 | g║ | g⊥ | A║ | A⊥ | B |
|---|
| V-meso TiO2 | air-exposed | 1.928 | 1.985 | −0.0174 | −0.0068 | 4.3 |
| evacuated | 1.925 | 1.985 | −0.0174 | −0.0071 | 4.5 |
| V/meso TiO2 | air-exposed | 1.925 | 1.972 | −0.0171 | −0.0068 | 2.6 |
| evacuated | 1.938 | 1.993 | −0.0172 | −0.0073 | 6.9 |
| B=(g║−ge)/(g⊥−ge) |
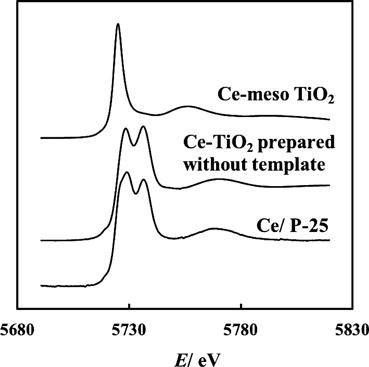
Moを担持メソ細孔性チタニアは,P-25担持触媒と比較して配位数や酸化数に大きな変化が見出せない29)。一方,図3に示すように,Ce L3吸収端XANESからは,直接合成したCe-meso TiO2ではCeの酸化数はⅢで,Ce/P-25ではⅣ価Ceが優勢である42)。エタノール-酸素反応では,前者が選択的にアセトアルデヒドを与えるのに対し,後者はエチレンとCO2を与える。さらに酸化処理を行うと,前者では酢酸への選択性が酸化温度とともに増大するのに対し,後者ではほとんど選択性が変わらない(表2)。
表2 メソ細孔性チタニア担持Ce触媒とP-25担持Ce触媒上のエタノール−酸素反応| 473 K焼成 | 転化率 | 選択率[%] |
|---|
| [%] | CH2=CH2 | CH3CHO | CH3COOH | CO2 |
|---|
| Ce-meso TiO2 | 77 | 5.2 | 93 | 0.1 | 1.7 |
| Ce/P-25 573 K焼成 | 96 | 52 | 1.9 | 0 | 46 |
| Ce-meso TiO2 | 69 | 15 | 31 | 53 | 1 |
| Ce/P-25 673 K焼成 | 99 | 64 | 0 | 0 | 3.6 |
| Ce-meso TiO2 | 60 | 5 | 26 | 68 | 1 |
| Ce/P-25 | 99 | 55 | 0 | 0 | 45 |
| Catalyst: 1.6 wt% Ce-meso TiO2 and 1.0 wt% Ce/P-25. p(C2H5OH)=4.32×103 Pa, p(O2)=4.85×104 Pa, p(Ar)=balance, T=423 K. |
このようにVとCeなどの易還元性の触媒成分の例では,非晶質メソ細孔性チタニアは低価数化学種を安定化させる効果が顕著であることが明らかになった。
還元されたチタニアに窒素分子が化学吸着することは古くから知られているが43),非晶質メソ細孔性チタニアを有機チタン化合物で処理後,窒素と水素に接触させると室温で表面にアンモニアが生成するという報告がある44)。
メソ細孔性ルチルではLiの挿入反応が研究されている。n-デシル硫酸11),Triton X-10022),SBA-15やKIT-624)を型剤に調製されたメソ細孔性ルチルはメソ構造が異なるものの,充放電特性は似ており,LiTiO2→Li++TiO2+eの反応に匹敵するLiの挿入-脱離が起きるが,充放電を反復すると容量は減少し,140 mA h/g程度になる。
ブロック共重合体を型剤にして,メソ細孔性チタニア薄層をITO電極上に生成させることも可能である。このように作成したメソ細孔性チタニア膜はphotovoltaic cellの電極に利用できる。また色素増感型太陽電池の電極はそもそもメソ細孔性のチタニア微結晶子集合体から成り46),メソ細孔性チタニアの重要な応用となっている。この場合も細孔径を均一化し,さらに細孔径を変化させて電極を調製すれば,色素分子の効率よい配置や反応中の物質拡散の効果を解明し,電池性能を向上するために重要な手段となりうる47)。