現在,燃料や化学原料の多様化を目的として,天然ガスや石炭,バイオマスと言った非石油系資源から,高付加価値な炭化水素に転換する技術に関する研究が精力的に行われている。その中でゼオライトを用いたメタノールやエタノールの転換反応によるプロピレン製造は注目を集めている。
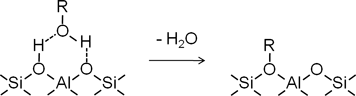
ゼオライトに室温でメタノールやエタノールを導入すると,酸性OH基上に水素結合を形成して吸着する。その後,昇温することで酸性OH基とアルコールの間で脱水して表面アルコキシ種を形成することが核磁気共鳴法(NMR)1,2)や赤外分光法(IR)3–5)を用いた観察から報告されている(スキーム1)。当グループではIR法によるゼオライト表面の観察と質量分析(MS)法による脱離成分の測定から,表面エトキシ種からエチレンが生成する際の活性化エネルギーを算出した。その結果,モルデナイト,H-ZSM-5,そしてフェリエライトの順で,細孔径が小さくなるほど活性化エネルギーが高くなることを見出した6)。本稿では,IR法を用いたメトキシ種とエチレンの反応を検討した結果を中心に紹介する。
H-ZSM-5を用いたメタノールからガソリン成分を合成するMethanol to Hydrocarbons(MTHC)は,1976年にMobil社のMeiselらによって報告された8)。この研究は第一次オイルショック後に発表されたため,大きな注目を集めた。日本では1980年から始まったC1化学プロジェクトの一端としてゼオライトを用いたメタノール転換反応の研究が行われた。近年においてもプロピレン需要の増加などから低級オレフィンの収率の向上を図ったMethanol to Olefin(MTO)が注目を集めている。
MTHCやMTOは反応生成物分布に対する接触時間依存性からスキーム2の経路で反応が進行すると考えられている9)。まず,メタノールの脱水反応によりジメチルエーテルが生成する。次にメタノールとジメチルエーテルの平衡状態から低級オレフィンが生成する。その後,生成したオレフィンがメチル化や重合反応などを通して炭素鎖成長を起こしたり,環化や水素移行により芳香族やコーク成分が生成すると考えられている。また近年はハイドロカーボンプール機構10,11)による反応の解釈が広く行われている。これはポリメチルベンゼン類などのハイドロカーボンプール種が,メタノールやジメチルエーテルによるメチル化を介して,低級オレフィンの生成とハイドロカーボン種の回復を繰り返すものである。しかし,メタノールやジメチルエーテルからオレフィンが生成する際の初期炭素-炭素結合の形成など,詳細な反応機構は未だ解明されていない。
近年,HungerらはNMRを用いてメトキシ種の反応性について検討を行ってきた2,12,13)。彼らはハイドロカーボンプール種に関わるトルエンがメトキシ種によってメチル化されてキシレンが生成することを見出した。そこで,我々はメタノール転換反応の素反応の1つであると考えられるメトキシ種によるエチレンのメチル化に着目して,IRを用いたメトキシ種の反応性の検討14)を行った。
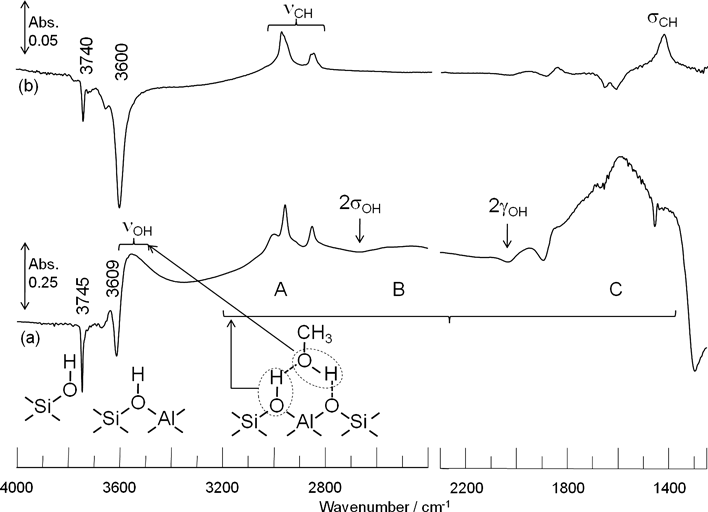
図1はH-ZSM-5(JRC-Z5-90H)に25°C (a) と300°C(b)でメタノールを導入して排気した時に観測されるスペクトルである。このスペクトルはメタノール導入前のスペクトルを差し引いた差スペクトルとなっている。以後,IRスペクトルは前処理後のスペクトルを差し引いたものを示す。25°Cのスペクトルで3744 cm−1と3609 cm−1の逆ピークはそれぞれ孤立した状態のシラノールと酸性OH基の減少を示している。これはシラノールと酸性OH基がメタノールと水素結合を形成するためである。また,吸着メタノールに帰属される吸収は,OH伸縮振動が3500 cm−1付近,CH伸縮振動が3000〜2800 cm−1に観測された。
水素結合した酸性OH 基は低波数側にシフトして,〜2900 cm−1と〜2400 cm−1と〜1600 cm−1にABCバンドと呼ばれる特徴的なバンドを形成する15–17)。このABCバンドは低波数側にシフトした酸性OH基のOH伸縮振動が酸性OH 基の面内変角振動および面外変角振動の倍音とフェルミ共鳴することにより,1つのブロードな振動が二つの逆ピークによって分割されて形成されたものである。また,メタノールのCH変角振動はCバンドと重なるため,明確なピークとなって現れない。一方,300°Cでメタノールを導入した場合,メタノールのOH基伸縮振動とABCバンドが観測されなかった(図1b)。しかし,3740 cm−1と3600 cm−1に孤立シラノールと孤立酸性OH基の減少に基づく逆ピークと3000〜2800 cm−1のCH 伸縮振動と1460 cm−1のCH変角振動に基づくピークが観測されたままである。したがって,水素結合したメタノールが脱水して,シラノールと酸性OH基上にメトキシ種が生成したことを確認した。メタノールの導入と排気を繰り返した結果,メトキシ種の酸性OH基に対する最大被覆率は約40%であり,300°Cで60分間真空排気しても酸性OH基上のメトキシ種は減少しなかった。このことから,300°Cではメトキシ種の分解やメトキシ種同士の反応は起こらないことがわかった。次にメトキシ種の反応性を検討した。
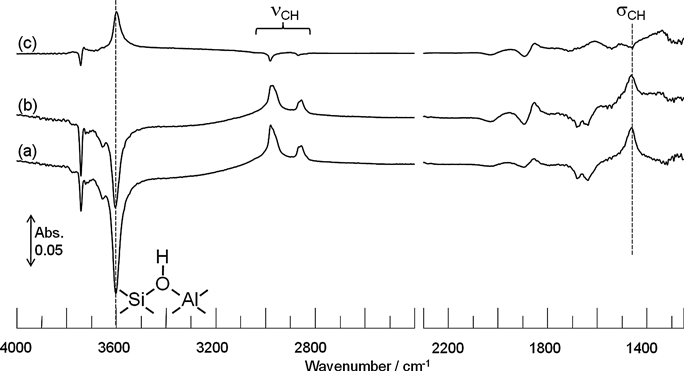
300°Cで生成したメトキシ種に対して250°Cでエチレン(メトキシ種の25% モル)を導入したときに観測されたIRスペクトルの経時変化を図2に示す。導入30分後と導入前の差スペクトル(図2c)から酸性OH基の回復に基づくピークとともに,CH伸縮および変角振動の減少に基づく逆ピークが観察された。メトキシ種は真空中では安定に存在することから,酸性OH基上のメトキシ種がエチレンと反応して減少したと考えられる。この時の気相成分の分析結果を表1に示す。この結果から導入5分後ではプロピレンのみ生成することがわかった。一方,メトキシ種が存在しないH-ZSM-5に対して,エチレンを単独で導入した場合には60分経っても反応が起こらなかった。したがって,メトキシ種とエチレンからプロピレンが生成したことが分かった。また,時間経過とともに生成物の炭素数が増加していくことから生成したオレフィンが逐次反応によって炭素鎖成長したと考えられる。
表1 H-ZSM-5上のメトキシ種とエチレンの反応における生成物分布a)| 反応時間/min | エチレン転化率/% | 収率/mol% |
|---|
| C3= | C4 | C5-C6 |
|---|
| 5 | 0.8 | 0.8 | 0 | 0 |
| 10 | 2.3 | 1.9 | 0.4 | 0 |
| 30 | 12.3 | 8.2 | 3.4 | 0.8 |
| 60b) | 0 | — | — | — |
| a)触媒:60 m; エチレン;15 Pa; 反応温度:250°C.b)メトキシ種を形成させていないH-ZSM-5を用いた場合. |
メトキシ種とエチレンからプロピレンが生成することを確認するため,13C-メタノールを用いて,13C- メトキシ種を形成させてエチレンと同様の条件で反応させた。MSスペクトルから反応初期に生成したプロピレンには13Cが一つ含まれており,13C-メトキシ種とエチレンからの生成を支持する結果が得られた。また,導入60分後のエチレンに含まれている13Cの存在比は自然存在比と変わらないことから,13C-メトキシ種同士からエチレンが生成しないことも確認された。以上のことから,エチレンはエチレン同士よりもメトキシ種によるメチル化によるプロピレン生成の方が進行しやすいことがわかった。Kolboeらも流通系反応装置を用いた検討から,メタノール転換反応中におけるエチレンの重合反応の影響は小さく,メタノールによるエチレンのメチル化の寄与の方が大きいことを報告している18)。
H-ZSM-5上のメトキシ種とエチレンの反応における表面メトキシ種と気相エチレンのそれぞれの反応次数を検討した。反応条件は300°C,10分間に固定し,メトキシ種の転化率とエチレンの転化率を測定した。
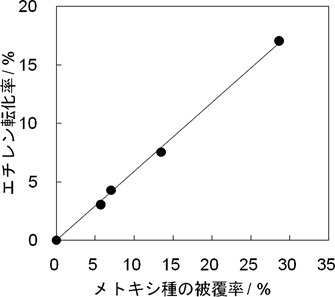
まず,酸性OH基に対するメトキシ種の被覆率依存性を検討するため,異なる被覆率のメトキシ種に対してエチレンの導入圧を一定(15 Pa)にして反応させた。横軸にメトキシ種の酸性OH基に対する被覆率,縦軸にエチレンの転化率をとると,傾きが正の直線が得られた(図3)。したがって,エチレンの転化反応はメトキシ種の量に対して1次で進行することがわかった。
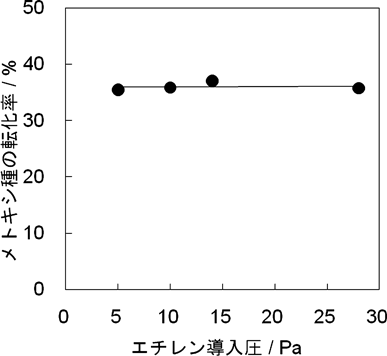
次に,エチレンの導入圧依存性を検討するため,被覆率が35%のメトキシ種に対してエチレンの導入圧を変化させて反応させた。反応前のメトキシ種の被覆率を100%としてメトキシ種の減少率をメトキシ種の転化率とすると,メトキシ種の転化率はエチレンの導入圧に対して傾き0の直線で得られた(図4)。したがって,メトキシ種の転化率はエチレンの導入圧に依存しないことがわかった。以上のことから,メトキシ種とエチレンの反応はメトキシ種の被覆率にのみ依存することがわかった。
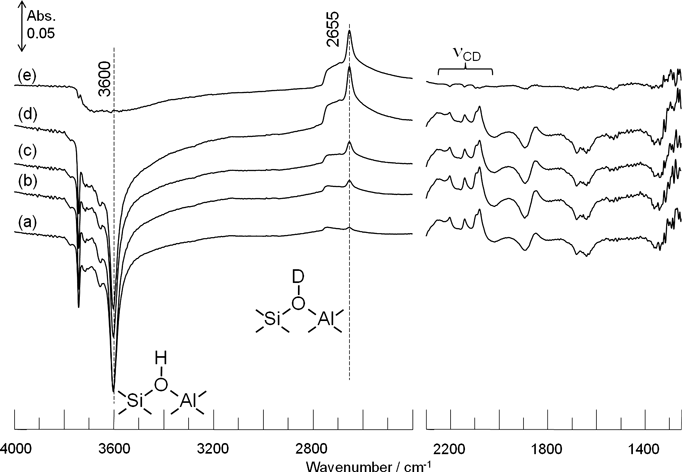
反応メカニズムをより詳細に検討するために,[d3]-メタノールを用いて300°CでH-ZSM-5上に[d3]-メトキシ種を形成させた。この[d3]-メトキシ種に図2の実験と同様に,反応温度250°Cでエチレンを導入した。図5aに示したように酸性OH基の減少による逆ピークとともにCD 伸縮振動が2300〜2000 cm−1に観測され,H-ZSM-5上に[d3]-メトキシ種が形成したことを確認した。また,CD変角振動は同位体シフトによる低波数シフトのため,観測することが出来なかった。[d3]-メトキシ種に対してエチレンを導入すると,酸性OD基に起因する2655 cm−1のピークが現れ,増加していく様子が観測された。反応20分後と反応前の差スペクトル(図5e)をとると,CD伸縮振動の減少に基づく逆ピークと酸性OD基の増加に基づくピークが確認された。一方,3600 cm−1の酸性OH基は導入前後で変化しないことがわかる。これはプロピレンが生成する際に,エチレンの水素ではなく[d3]-メトキシ種の重水素が選択的に表面に残り,酸性OD基を生成することを示している。したがって,[d3]-メトキシ種とエチレンからプロピレンが生成するときに[d3]-メトキシ種はメチル基(CD3-)としてではなく,メチレン基(CD2-)としてエチレンに導入されることがわかった。
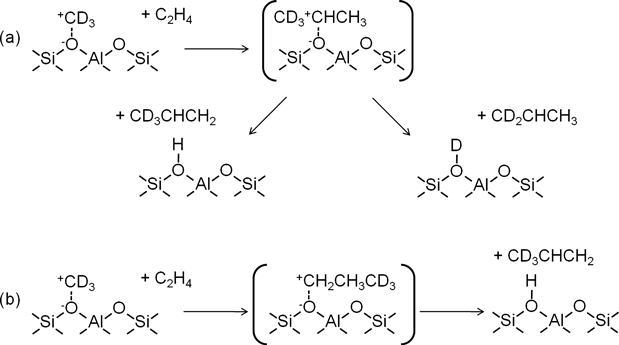
ゼオライト表面に形成されるアルコキシ種は理論計算から遷移状態としてカルベニウムイオンを経由して反応することが報告されている19,20)。また,小野らは酸性OH基のプロトンが表面を移動するようにメトキシ種からメチルカチオンが生成して移動すると提唱した3,21)。このメトキシ種からのメチルカチオンの生成はDFT計算によってモデル化されている22,23)。カルベニウムイオン機構で[d3]-メトキシ種とエチレンの反応が進行すると考えると,[d3]-メトキシ種が[d3]-メチルカチオンとしてエチレンと反応し,2-プロピルカチオンを形成すると考えられる。この2-プロピルカチオンからプロピレンと酸性水酸基を生成する場合,2つの経路が考えられる(スキーム3a)。CD3CH=CH2が脱離して酸性OH基が生成する経路と,CD2=CHCH3が脱離して酸性OD基が生成する経路である。つまり,この2-プロピルカチオンを経由する経路では酸性OH基と酸性OD基の両方が生成する。これは同位体効果を考慮しても酸性OH 基が観測されなかったことと矛盾する。したがって,2-プロピルカチオンを経由した反応は進行していないと考えられる。
一方で最近,理論計算からメトキシ種とエチレンから1-プロピルカチオンが生成することが提唱されている23)。この機構では[d3]-メトキシ種とエチレンから+CH2CH2CD3の1-プロピルカチオンを形成後,CH2=CHCD3が脱離して酸性OH基が生成する(スキーム3b)。このように1-プロピルカチオンを経由して反応が進行する場合,酸性OH基が生成するはずである。これら2 つの異なる反応経路を仮定しても,メトキシ種とエチレンからプロピレンを生成する反応はカルベニウムイオン機構で進行していないと考えられる。
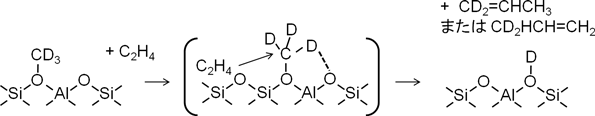
HungerらはNMRを用いた反応観察から,メトキシ種のC–H結合が格子酸素によって活性化されたカルベン的な反応中間体の形成を提唱した12,13)。このカルベン的な中間体を経由して反応が進行すると仮定すると,格子酸素によってC–D結合が活性化された[d3]-メトキシ種と吸着エチレンが反応してプロピレンが生成すると考えられる(スキーム4)。この場合,プロピレンが脱離する時には酸性OD基だけが生成する。これはIR法による直接観察による結果とよく一致するため,H-ZSM-5上のメトキシ種とエチレンからプロピレンを生成する反応はカルベン的な中間体を経由して協奏的に反応が進行すると考えられる。また,Si/Al比の異なるHZSM-5(Si/Al=25, 150)やトポロジーの異なるゼオライト(FAU, MOR, CHA)を用いた場合にも同様な結果を得た。したがって,ゼオライト上のメトキシ種とエチレンの反応はカルベン的な中間体を経由して反応が進行することが示唆された。
同位体を用いてH-ZSM-5上のメトキシ種とエチレンの反応を検討した結果,メトキシ種がカルベン的な反応中間体を経由し,エチレンと反応してプロピレンが生成することを見出した。また,この反応は気相エチレン圧には0次で,表面メトキシ種の量に1次であることから,格子酸素によるメトキシ種のC–H結合の活性化が律速段階であると考えられる。今後,さらにメタノール転換反応の機構について詳細に検討していきたいと考えている。
引用文献References
1) W. Wang, M. Seiler and M. Hunger, J. Phys. Chem. B, 105, 12553 (2001).
2) W. Wang, J. Jiao, S. S. Ray and M. Hunger, Chem. Phys. Chem., 6, 1467 (2005).
3) Y. Ono and T. Mori, J. Chem. Soc. Faraday Trans. 1, 77, 2209 (1981).
4) T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 109, 5076 (1987).
5) J. N. Kondo, K. Ito, E. Yoda, F. Wakabayashi and K. Domen, J. Phys. Chem. B, 109, 10969 (2005).
6) J. N. Kondo, D. Nishioka, H. Yamazaki, J. Kubota, K. Domen and T. Tatsumi, J. Phys. Chem. C, 114, 20107 (2010).
7) 触媒学会編,触媒便覧,講談社 (2008).
8) S.L. Meisel, J. P. McCullough, C. H. Lechthaler, and P. B. Weisz, Chemtech, 6, 86 (1976).
9) C. D. Chang and A. J. Silvestri, J. Catal., 47, 249 (1977).
10) I. M. Dahl and S. Kolboe, Catal. Lett., 20, 329 (1993).
11) J. F. Haw, W. Song, D. M. Marcus and J. Nicholas, Acc. Chem. Res., 36, 317 (2003).
12) W.Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 125, 15260 (2003).
13) W. Wang and M. Hunger, Acc. Chem. Res., 41, 895 (2008).
14) H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Angew. Chem. Int. Ed., 50, 1853 (2011).
15) A. G. Pelmenschikov, J. H. M. C. van Wolput and R. A. van Santen, J. Phys. Chem., 99, 3612 (1995).
16) A. Zecchina, S. Bordiga, G. Spoto, D. Scarano and F. Geobaldo, J. Chem. Soc. Faraday Trans., 92, 4863 (1996).
17) C. Paz, S. Bordiga, C. Lamdetri, M. Salvalaggio, A. Zecchina and G. Bellussi, J. Phys. Chem. B, 101, 4740 (1997).
18) S. Svelle, P. O. Ronning and S. Kolboe, J. Catal., 224, 115 (2004).
19) V. B. Kazanski, Catal. Today, 51, 419 (1999).
20) P. E. Sinclair, Ade Vries, P. Sherwood, R. C. A. Catlow and R. A. van Santen, J. Chem. Soc. Faraday Trans., 94, 3401 (1998).
21) T. Baba, T. Mori, Y. Ono and H. Sugihara, J. Phys. Chem. B, 102, 804 (1998).
22) C.M. Zicovich-Wilson, P. Viruela and A. Corma, J. Phys. Chem., 102, 12334 (1995).
23) T. Maihorn, B. Boekfa, J. Sirijaraensre, T. Nanok, M. Probst and J. Limtrakul, J. Phys. Chem. C, 113, 6654 (2009).