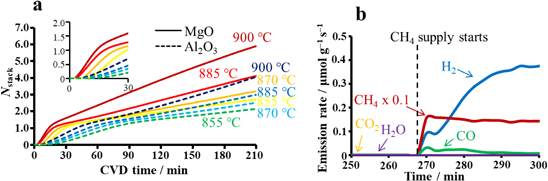
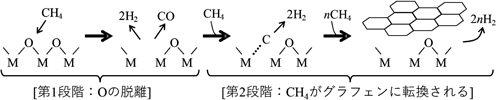
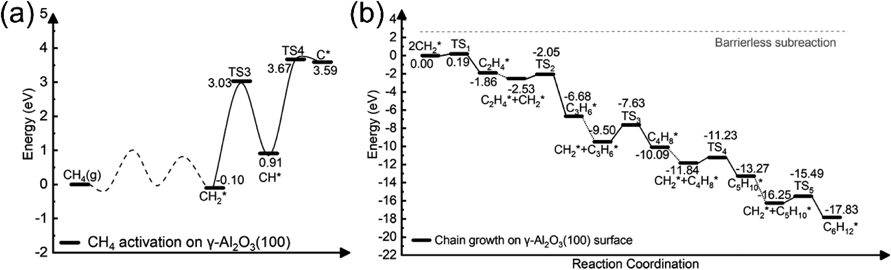
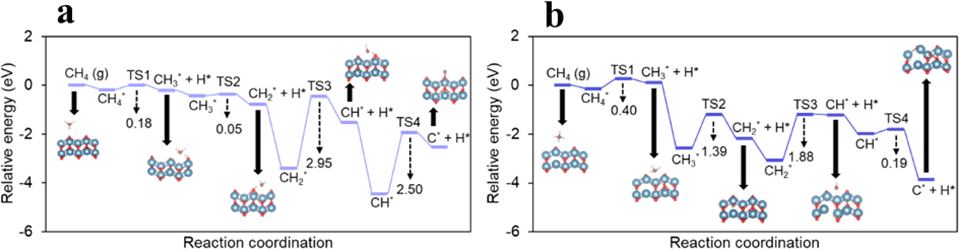
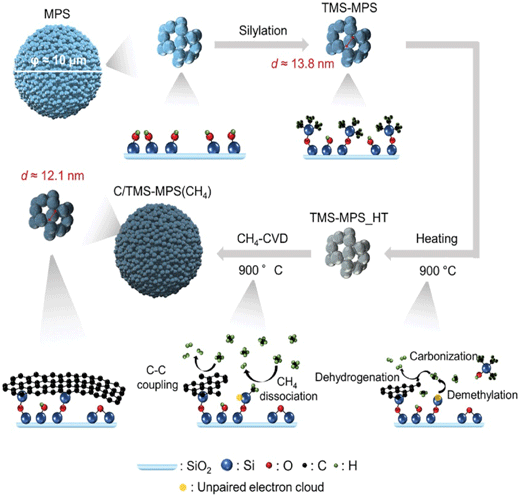
酸化物セラミックス上でのメタン活性化とグラフェン成長メカニズムMechanism of Methane Activation and Graphene Growth on Oxide Ceramics
1 東北大学多元物質科学研究所Institute of Multidisciplinary Research for Advanced Materials, Tohoku University ◇ 〒980–8577 宮城県仙台市青葉区片平2–1–1
2 東北大学材料科学高等研究所Advanced Institute for Materials Research (WPI-AIMR), Tohoku University ◇ 〒980–8577 宮城県仙台市青葉区片平2–1–1
受理日:2024年10月8日Accepted: October 8, 2024
発行日:2025年1月31日Published: January 31, 2025